臨床藥理學在創新藥研發中的最新進展*
2017-10-26 08:25:15胡蓓
世界科學技術-中醫藥現代化 2017年7期
胡 蓓
(北京協和醫院I期臨床研究室 北京 100032)
臨床藥理學在創新藥研發中的最新進展*
胡 蓓**
(北京協和醫院I期臨床研究室 北京 100032)
在傳統的“經驗描述”新藥開發模式下,創新藥研發無法綜合利用已有信息,開發效率低下,并且容易引發安全性隱患。因此,探索并建立更高效更安全的創新藥物臨床開發新模式和新機制尤為必要。近年來,FDA和EMEA陸續提出并踐行的以“知識綜合”為特征的藥物臨床研究模式就是其中的重要代表。它利用模型與模擬技術定量地綜合分析和預測創新藥在患者體內的暴露/效應關系及其影響因素,幫助在獲取足夠信息的同時,在臨床試驗中盡量減少所需受試者數量,并保障臨床試驗的安全性。
定量藥理學 模型化與仿真 PK/PD 樣本量
1 簡述
近年來,試驗設計方法和數據分析方法的變化是我國臨床藥理學領域重要的研究進展。
1.1 臨床藥理學在創新藥研發中的重要性
從ICH-E8臨床試驗的一般性考慮可知,臨床藥理學(Clinical Pharmacology,CP)在創新藥早期臨床研發中所占比重很大(圖1)。從圖1顯示的內容可知,臨床藥理學研究貫穿整個藥物研發過程。在研發早期進行的次數比較多(該階段的顏色比較深);在臨床研究過程中根據研究需要還會進行相應的臨床藥物學研究,但相對研究早期研究階段,研究數目有所減少(該階段的顏色變淺)。每一個研究都需根據其研究目的制定研究計劃、設計研究方案,獲得與研究目的相關的研究數據。通過數據的分析和挖掘,總結成臨床試驗報告提交給藥政管理部門和申辦者,用以指導下一步的臨床試驗。值得注意的是,有時在治療作用探索在早期臨床研究階段就可以進行,此時也可能把臨床藥理學的研究內容和初步的療效評估相結合。
近年來FDA出臺了鼓勵新藥研發的管理措施,包括突破性治療藥物的批準。如果在早期就能發現明顯治療作用,在未完成III期臨床試驗的情況下,有的創新藥也可以獲得突破性治療藥物的批準,有條件的上市。由于臨床藥理學研究在藥物研發早期比重較高,這使得它在這類創新藥的研發過程中的作用很突出。2015年,FDA一共批準了45個新藥,其中不少藥物獲得了突破性治療藥物批準,這些藥物上市前研究中臨床藥理學所占比重是非常大的。所以,臨床藥理學研究在創新藥研發中的作用越來越重要。
另外,臨床藥理學的重要性還體現在我們需要在安全的前提下,盡快地推進研究,盡快地獲得數據,盡快地獲得研究結果,盡早地幫研發者做出決策。臨床藥理學能更快地向前推進的主要原因是臨床藥理學研究是都是小樣本量研究,比大樣本量臨床試驗獲得數據的速度更快,因此能更快地得出結論和做出決策。
1.2 臨床藥理學的研究目的及核心研究內容
在早期的臨床藥理學試驗中,從動物到人體,需要預測人對藥物的耐受性和藥物的安全性。2016年法國“雷恩”事件之后業界對安全性又有了新的考慮,事實上安全性問題一直都有新的熱點值得討論。臨床藥理學研究人員是在現有的認識之下,在可接受的安全性下,預測人體對在研新藥的最大耐受劑量,包括單劑給藥和多劑給藥情況。臨床藥理學研究可以在全劑量范圍之內表征劑量限制性的、跟暴露量相關的不良效應;如果有合適的生物標志物,還可以在這個范圍內表征藥代/藥效關系。如果生物標志物和藥物作用機制、臨床獲益相關,臨床藥理學研究還可以獲得機制驗證或概念驗證的研究結果。
因此,劑量-暴露-效應關系是臨床藥理學研究的核心內容。
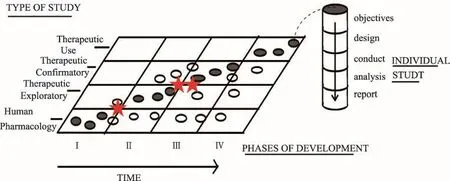
圖1 臨床藥理學在創新藥早期臨床研發中所占比重
2 中國臨床藥理學的最新進展
2.1 研究設計的變化
2.1.1 高內涵研究(Umbrella Study Design,USD)
在臨床藥理學試驗中應盡可能地研究藥物暴露量和效應之間的關系,包括生物標志物信息和安全性信號,在此基礎上評價藥物和機體的相互作用。
2.1.2 多種研究工具
臨床藥理學研究者可采用多種研究工具,主要包括:(1)在劑量遞增的過程中,除了用異速生長放大法從動物劑量換算到人體的用藥劑量外,還可使用其他方法,如PK/PD暴露量指導的劑量遞增,實時地獲得每個劑量組各個受試者的藥物濃度、生物標志物的暴露量,根據動物實驗暴露量與安全性、有效性的關系,在藥物劑量遞增的情況下重新評估人類給藥后的安全性,再指導藥物劑量遞增。這樣可以使研究更有效率,而且能在最大程度上保證受試者的安全。
另外,還可以通過“模型和仿真”的方法來總結非臨床試驗數據之間的規律,關注這些規律在人體上是否能夠重現,并由此從動物數據預測人類體內的藥物暴露量。再根據預測設計試驗,獲得新研究數據,用新的數據確認以往的規律總結、優化模型;再運用新模型設計試驗來進行新的數據收集,這種基于模型預測的試驗方案設計是一個“學習-確認-再學習-再確認”向前迭代推進的過程。這種研究策略可以幫助研究者更快、更有效率地將臨床試驗向前推進,使每一次研究的信息被充分利用。
2.1.3 基于臨床藥理學數據的變異確定給藥方案
試驗所獲得的臨床藥理學數據存在個體變異。研究者可根據變異的大小及變異與治療窗的關系,在安全性允許的前提下,以最大限度地使得更多的患者藥物暴露能夠保持在治療窗之內為目標,通過模型預測合適的給藥方案,并在臨床試驗中加以驗證或進一步調整,使得研究者可以盡早確定II、III期臨床研究的給藥方案,大大提高研究效率。這類臨床藥理學研究可以采用群體分析法研究參數的變異的參數及變異來源。
2.2 數據分析方法的變化
2.2.1 正確表征研究樣本的PK/PD特征
通過PK/PD參數數據的典型值及變異(分布)的研究,分析協變量,可以厘清對個體差異貢獻較大的個體因素(協變量),并分析這些影響因素與治療窗的關系,估計劑量調整的必要性。因此近年來臨床藥理學研究的一個新進展是PK/PD參數表征方法的變化。
2.2.2 從統計學的角度評判研究結果的可靠性
所有的臨床試驗數據均可溯源、數據質量有保障只是臨床試驗研究結果可靠的前提之一。另一個判斷臨床試驗研究結果是否可靠的標準是評估這一結果是否為大概率事件,即研究結果是否可以在同樣試驗條件下可重現。由于臨床藥理學試驗經常是小樣本研究,其研究結果能否正確表征研究藥物在更大人群中的PKPD特征,在很大程度上依賴于該研究藥物PKPD參數在人群中的變異。變異越小,研究結果依賴于抽樣的程度就越低,研究結果可以重現的概率越高,該數據就越可靠,反之亦然。既然臨床藥理學研究結果的可靠性與變異和樣本量相關,那么如何根據臨床試驗結果的變異與樣本量的關系評判研究結果的可靠性呢?研究者、申辦者及審評者都需要考慮評判臨床藥理學研究結果是否可靠的標準,并形成共識。當然增加研究結果的可靠性的根本方法就是增加樣本量,但這與臨床藥理學的小樣本研究的現實并不相符,在很多情況下并不具有可操作性。為此我們提出兩種解決方案:①適當擴大入組人數,增加樣本量;②有條件的將同類研究合并分析(多個研究互相印證)。在多個試驗的結果確與藥物的某固有性質相關時,此法有利于通過合并分析增加樣本量,提高研究結果可重現的概率,是較好的解決方案。
2.3 需要事先制定研發策略
對于不值得繼續開發的藥物通過臨床藥理學研究盡早發現其缺陷,盡快終止其進一步的臨床開發,同樣可以定義為成功的研究。如果在試驗中遇到非預期的試驗結果,研究者需要要有能力從中盡可能多地學習,了解其中的原因。使用定量藥理學技術能在研發的過程中幫助確定合適的劑量并幫助設計試驗。以往數據顯示,如果研發早期的臨床藥理學試驗做得好的話,40%的新藥可在早期臨床試驗階段終止開發,是用非臨床數據預測臨床數據時有相當大的不確定性導致的,這是藥物研發風險最高的階段。隨著非臨床研究水平和跨種屬預測水平的上升,臨床研究的失敗率將會下降。經驗表明,包括臨床醫生、臨床藥理學研究者在內的新藥研發各個環節的專業人員跨學科合作,盡早制定以臨床為導向的非臨床和臨床研發策略,可以有效地提高研發效率,降低臨床研發失敗率。
2.4 早期臨床藥理學研究的強項與弱項
早期臨床藥理學研究強項包括:①精準:能準確的測定藥物的暴露量/濃度、生物標志物的變化、暴露量和效應之間的關系(PK/PD)并初步研究人體生物轉化等;②到位:透過現象(濃度的變化)揭示本質(藥物與機體的相互作用)等。早期臨床藥理學研究的弱項主要包括:①樣本量小,結果的代表性受質疑(人體的個體間變異通常比動物大);②由生物標志物變化所表征的藥效學不一定與臨床結局直接相關。
針對以上弱項臨床藥理學研究者提出了兩個解決方案:①研究的樣本量小,統計效能不足問題可以用安慰劑對照來突顯藥物和安全性信號之間的關系,具體辦法是研究暴露量和安全性信號的關系,如果兩者相關可以幫助確定藥物與安全性之間的量效關系;②當小樣本研究數據的變異較大時,可有條件地將多個研究合并分析,相互印證。
數據的變異很小時,抽樣對結果的影響較小,研究結果可靠;當多個研究、多次抽樣獲得的結果重現性很強時,小樣本研究的結果亦是可靠的。但如果數據變異較大,而抽樣量較少時,則可能出現問題。例如研究高中一年級男生的身高分布時,由于這個年齡段的男生正處于青春發育期,身高的變異會比較大。如果抽取的樣本量較小,即只測定少部分個體的身高,并以此對高中一年級男生總體的身高分布進行估計就可能會由于抽樣誤差產生對總體估計的偏差。例如抽樣時恰巧大都抽中了高個子,獲得的平均值可高于真實的平均值;相反如大都抽中了矮個子,均值就可能會低于真實的平均值,且測量結果重現的概率也較低。因而,數據變異大時,小樣本研究結果便可能不可靠。假設同類研究不止一個,研究的樣本都來自于同一個總體,那么就有條件合并在一起進行分析。這樣由于樣本量的增加,研究結果受抽樣影響的程度就會變小,結果就更能反映總體的分布特征,得出更準確的結果,也就是多個研究可以相互印證的結果。
圖2是入組人數與研究結果可以重現的把握度之間關系的示意圖。橫坐標是入組的人數,縱坐標是重做該試驗時結果可重現的把握度。如果研究對象的個體間變異是20%(綠線),要使研究重現的把握度高于80%的話,需要的樣本量約10-20例。如果研究對象個體間差異小于10%(藍線),哪怕樣本量僅有幾例,都有接近100%的把握重現原來的測定結果。當個體內的變異高達50%時,要使研究重現的把握度高于80%的話,需要的樣本量可高達70-80例。在這種個體間變異的情況下,如果進行的臨床藥理學試驗是20例以下的小樣本研究的話,則所得研究結果可以重現的把握度就會很低,即結果不太可靠。
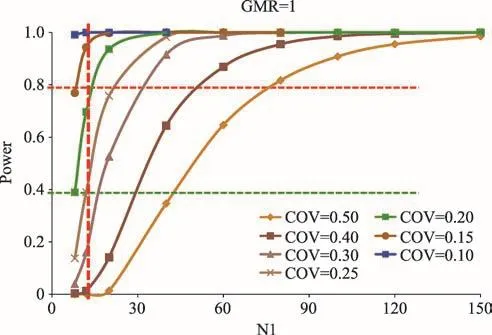
圖2 樣本量和變異對結果可靠性的影響
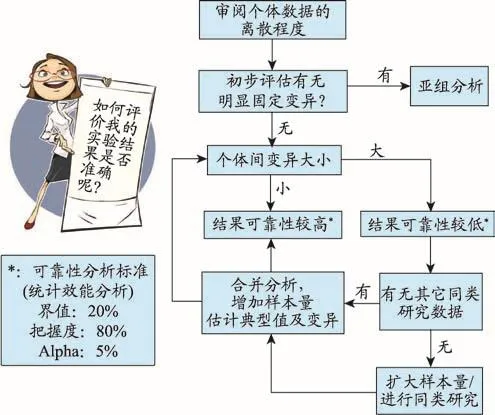
圖3 對早期臨床藥理學研究結果可靠性判斷的思路圖
研究結果可以用描述性統計法表述,也可以采用群體研究法用典型值及概率分布表述。描述性統計法的特點是關注均值,研究結果受抽樣影響較大,結果易受極值的影響;群體研究法通過研究變異關注研究人群全體,研究結果由于樣本量大受抽樣影響較小,因此群體研究法更可能得到對研究對象全體的正確評估。
2.5 對研究結果準確性判斷的思路圖
如圖3所示,第一步需要審閱研究數據集的離散程度,并評估有無明顯的固定變異。如果沒有就進行個體間變異大小的判斷,如果有就進行亞組分析。當數據個體間變異較小時,可預判研究可靠性較高。相反,變異大且樣本量小時,則可預判研究結果可靠性較低。一旦預判結果可靠性較低時,可參考其它同類研究數據,如果可以合并分析,則樣本量增加,結果是對典型值及變異的估計變得較為準確;如果沒有同類研究,就只能擴大樣本量重復試驗或者進行同類研究后再合并數據進行分析。通過結果準確性判斷過程,使研究者、申辦者和審評者對研究結果是否可靠有基本的估計,做到心中有數。俗話說,八九不離十,評判時可提前設定好兩次研究結果間的差異值和重現結果的把握度,例如差異設在正負20%之內,把握度設在80%。
3 創新藥早期臨床試驗設計要點
3.1 小樣本研究
創新藥早期臨床藥理學研究的特點是樣本量小,可探索新藥的耐受性和安全性,但不能證實安全性。可以用安慰劑對照來幫助厘清安全性信號與試驗藥物之間的關系。由于樣本量小,統計效能往往不足,但多數情況下可以進行多個同類研究,研究結果互相印證,以增加研究結果的可靠性。
3.2 首次人體試驗(FIH)
FIH研究中如能安全地達到最大耐受劑量,則可全面表征在研藥物的臨床藥理學特性。可在全劑量范圍內初步評估藥物的安全性和耐受性;評估藥代動力學(ADME)特征;初步評估暴露量與生物標志物(如果有)的關系;初步評估暴露量與劑量限制性毒性的關系;初步觀察試驗藥物在人體的生物轉化特點。為此,在安全性不妥協的前提下,應避免起始劑量過低或遞增太慢以至于無法獲得有效信息,應避免劑量遞增終止過早。基于跨種屬外推預測人類的PK、PK/PD設計臨床試驗方案,并輔以實時PK、PK/PD監測及密切地臨床安全性觀察,及時根據臨床試驗新信息及時調整試驗方案,有助于更好地把握首次人體試驗的安全性,更好地保障受試者的安全,并大大提高研究效率。
3.3 小結
正如千人千面一樣,病各不同藥亦不同。實際上,無法用一個研發策略適應所有創新藥物的臨床開發。即使同一治療領域,不同藥物,研發策略也不相同,需要研究者掌握要領靈活運用不同的臨床藥理學研究方法。定量藥理學臨床藥理學領域內進展最快、最活躍的學科分支,也是一個橋梁學科。它通過模型化和仿真的研究模式,將生物學、基礎藥理學、藥效學、藥動學、毒理學、醫學、統計學等學科知識融會貫通,起到聯通藥物研發過程中的各個知識孤島的作用,它的應用可貫穿新藥研發的全過程。我國的定量藥理學研究基礎雖然比較薄弱,但近年來研究進展十分迅速。
1 Chen J,Liu D Y,Zheng X,et al.Relative contributions of the major human CYP450 to the metabolism of icotinib and its implication in prediction of drug--drug icotinib and CYP3A4 inhibitors/interaction between inducers using physiologically based pharmacokinetic modeling,Expert Opin.Drug Metab.Toxicol.11(6):857-868,2015
2 Chen X,Matsuzawa T,Hitsu E,et al.Modeling the dermatopharmacokinetic profile of two loxoprofen patches for bioequivalence confirmation,Int J Clin Pyarmacol Ther,53(5):189-191,2015
3 Liu D Y,Ma X F,Liu Y,et al.Quantitative prediction of human pharmacokinetics and pharmacodynamics of imigliptin,a novel DPP-4 inhibitor,using allometric scaling,IVIVE and PK/PD modeling methods,European Journal of Pharmaceutical Sciences,89:73-82,2016
4 Liu D Y,Yang H,Jiang J,et al,Pharmacokinetic and Pharmacodynamic Modeling Analysis of Intravenous Esomeprazole in Healthy Volunteers,Journal of Clinical Pharmacology,56(7):816-826,2016
Latest Progress of Clinical Pharmacology in Innovative Drug Development
Hu Pei
(Phase I Clinical Research Office,Peking Union Medical College Hospital,Beijing 100032,China)
Under the traditional“empirical trial design”,innovative drug development cannot make comprehensive use of the existing information from previous studies.The development efficiency is low.And it is easy to cause safety issues.Therefore,it is particularly necessary to explore and establish new models,more efficient and safer strategies for the clinical development of innovative drugs.In recent years,the“knowledgeable”clinical research strategy for new drug has been proposed and practiced by the FDA and EMEA.It employed the model and the simulation technology to quantitatively analyze and predict exposure/response relationships of innovative drug and its influence factors in patients.It helped in obtaining sufficient information from clinical data at the same time,to minimize the required number of subjects,and to ensure the safety of subjects in clinical trials.
Pharmacometrics,modeling and simulation,PK/PD,sample size
10.11842/wst.2017.07.004
R36
A
2017-05-02
修回日期:2017-06-28
* 國家“十二五”重大新藥創制專項(2012ZX09303006-002):自身免疫病、糖尿病及骨質疏松藥物新藥臨床評價研究技術平臺,課題負責人:胡蓓、張奉春。國家“十三五”重大新藥創制專項(2017ZX09304031-001):“創新藥物早期臨床藥理學評價技術平臺建設,負責人:趙維剛。
** 通訊作者:胡蓓,教授,主要研究方向:臨床藥理學和定量藥理學技術在新藥研發中的應用
(責任編輯:郭嫦娥,責任譯審:王 晶)
猜你喜歡
課堂內外·初中版(科學少年)(2023年10期)2023-12-10 00:43:06
全科護理(2022年10期)2022-12-26 21:19:15
現代儀器與醫療(2022年2期)2022-08-11 09:51:40
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
汽車工程師(2021年12期)2022-01-18 06:02:43
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
國際放射醫學核醫學雜志(2021年10期)2021-02-28 08:41:58
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
信息安全與通信保密(2016年3期)2016-08-23 01:23:46
