以轉氨酶升高為主的多種酰基輔酶A脫氫酶缺乏癥1例臨床表現及基因分析
2017-11-01 13:36:20譚艷芳歐陽文獻姜濤唐蓮蘇雨霞李雙杰
中國中西醫結合兒科學 2017年5期
譚艷芳, 歐陽文獻, 姜濤, 唐蓮, 蘇雨霞, 李雙杰
罕見病研究
以轉氨酶升高為主的多種酰基輔酶A脫氫酶缺乏癥1例臨床表現及基因分析
譚艷芳, 歐陽文獻, 姜濤, 唐蓮, 蘇雨霞, 李雙杰
目的研究1例以轉氨酶升高為主的多種酰基輔酶A脫氫酶缺乏癥患者臨床表現、實驗室檢查、肌肉活檢及基因突變情況分析,并進行文獻復習,為該病的早期診斷及治療提供依據。方法收集1例7歲3月男性患兒的臨床資料,采集患兒及父母血標本,采用二代基因測序檢測致病基因,腓腸肌穿刺活檢明確肌肉病變情況。結果患兒肌肉活檢電鏡結果示肌纖維內大量脂滴沉積。基因測序結果顯示患兒的ETFDH基因存在c.1773_1774 del AT p.(Cys592※)無義突變和c.389A>T p.(Asp130Val)錯義突變,考慮為復合雜合突變,患兒父母分別為攜帶者。結論臨床上有轉氨酶升高為主伴有心肌酶升高、運動障礙者,應盡早進行分子遺傳學檢查,有條件者行腓腸肌肌肉活檢術,可以為患兒家庭提供準確的遺傳咨詢和產前診斷。
多種酰基輔酶A脫氫酶缺乏癥; 脂質沉積性肌病; ETFDH基因; 兒童
多種酰基輔酶A脫氫酶缺乏癥(multiple aeyl-CoA dehydrogenase deficiency,MADD)是一種脂肪酸β氧化代謝障礙性常染色體隱性遺傳病[1]。臨床表現輕重不一,表現高度異質,易被誤診,新生兒發病病情兇險,不易被早期發現,常可導致猝死,早期診斷和合理治療可明顯改善預后。本研究通過疑難病例基因檢測確診1例以轉氨酶升高為主的MADD患兒,并對家系進行基因分析,現報告如下。
1 病例資料
患兒男性,年齡7歲3個月,因“乏力、行走不穩3月余,發現轉氨酶升高10余天”于2015年6月1日入院。患兒家屬訴近3個月來患兒無明顯誘因出現全身乏力,行走不穩,爬樓梯需攙扶,無發熱,無意識障礙、抽搐等,未予特殊處理。10余天前在某院檢查,轉氨酶及肌酸激酶、乳酸脫氫酶均升高,肌電圖示“肌源性損害電生理改變”。個人史及家族史無特殊。查體:體質量20 kg,肝脾無腫大,四肢肌張力正常,雙下肢肌力4級,雙側膝反射正常,余無其他陽性體征。實驗室檢查:肝功能、心肌酶結果見表1。
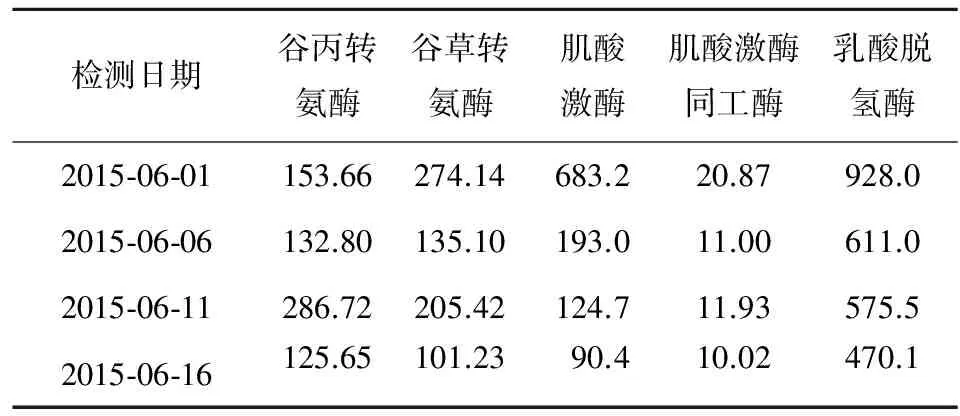
表1 患兒肝功能、心肌酶檢測結果(IU/L)
血脂:膽固醇5.71 mmol/L,高密度脂蛋白膽固醇3.28 mmol/L,低密度脂蛋白膽固醇2.52 mmol/L。銅藍蛋白、超敏肌鈣蛋白正常,降鈣素原、血沉、C反應蛋白正常。EBV-DNA及抗體陰性,甲、乙、丙、丁、戊肝抗體陰性。頭顱MRI:松果體囊腫可能性大。雙下肢肌電圖:肌肉收縮時部分肌肉呈不典型肌源性病損改變。血串聯質譜分析:顯示多種酰基肉堿增高,伴游離肉堿降低。尿液有機酸分析:顯示多種有機酸增高。心臟彩超:未見異常。
基因檢查方法:抽取患兒及父母靜脈血1~2 mL送檢,取200 μL全血,常規抽提血液基因組DNA,行脂肪酸氧化缺陷與肉堿循環缺陷相關基因共34個基因檢測。應用過柱法從外周血提取DNA,然后應用測序技術,對脂肪酸氧化缺陷與肉堿循環缺陷相關基因的外顯子編碼區進行直接測序,與參考序列進行比較,從而發現可能存在的基因突變。
基因測序結果:檢測到患兒一個致病突變和一個可疑致病突變,ETFDH第13外顯子,c.1773_1774 del AT p.(Cys592※)雜合致病突變(圖1a,見封三),第3外顯子,c.389A>T p.(Asp130Val)(圖1b,見封三),可疑雜合致病突變,故考慮復合雜合突變。父母特定位點基因分析,父親攜帶c.1773_1774 del AT p.(Cys592※)雜合突變(圖1c,見封三),母親該位點未見異常(圖1d,見封三);母親攜帶c.389A>T p.(Asp130Val)雜合突變(圖1e,見封三),父親該位點未見異常(圖1f,見封三)。
腓腸肌活檢標本光鏡結果:鏡下肌纖維結構部分清楚,可見橫紋結構,未見明顯炎細胞浸潤,肌纖維內脂滴沉積,糖原增加,見圖2。

圖2 多種酰基輔酶A脫氫酶缺乏癥患兒肌活檢標本光鏡結果
治療上給予患兒口服維生素B2每次20 mg,每日3次;輔酶Q10膠囊每次10 mg,每日3次;左卡尼汀口服液100 mg/(kg·d),并進行低脂、低蛋白及高碳水化合物的飲食指導。隨訪23個月,患兒無任何不適,復查肝功能、心肌酶、血脂均無異常。
2 討論
MADD是由于編碼線粒體的電子轉運黃素蛋白或電子轉運黃素蛋白-泛醌氧化還原酶,也稱電子轉運黃素蛋白脫氫酶(electron-transferring flavoprotein dehydrogenase,ETFDH)的基因發生突變所致的常染色體隱性遺傳性疾病[1]。是一種罕見疾病,國內報道少,而遲發型的病例報道則更少[2]。由于此病臨床表現多種多樣,并缺乏特異性的體征與癥狀,臨床診斷較為困難,易被延誤診斷,從而影響治療及預后。
ETFDH基因位于4q33,含13個外顯子。其發病機制為電子轉運黃素蛋白接受來自脂肪酸β氧化過程中多種脫氫酶產生的電子,轉運至線粒體內膜的ETFDH,并經由ETFDH所結合的泛醌轉運至呼吸鏈復合體Ⅲ,產生ATP為機體供能。如ETFDH缺陷,可引起呼吸鏈多種脫氫酶功能障礙,導致脂肪酸及能量代謝等受阻、紊亂,從而出現諸如大量脂質沉積、肌肉乏力、低酮性低血糖及代謝性酸中毒等臨床癥狀[3]。主要累及肢體近端肌肉,尤其是頸部屈肌和伸肌,嚴重時不能抬頭,少數出現呼吸困難或呼吸肌無力[4]。
MADD可分為3型,新生兒發作型根據伴或不伴有先天發育異常,分為Ⅰ型和Ⅱ型,遲發型又稱為Ⅲ型。新生兒發作型表現較重,常有低血糖腦病、肌張力低下、呼吸急促或呼吸困難、嚴重酸中毒等。而遲發型患兒在生后數周致成人均可發病[2],臨床表現無特異性,多隱匿起病,表現相對較輕,主要表現為間歇性肌無力,可累及軀干及四肢近端骨骼肌,也可出現心肌、肝臟損害。血串聯質譜可有短、中和長鏈酰基肉堿(C4~C18)的酰基肉堿升高,尿氣相質譜分析可有大量有機酸排出,主要是戊二酸和乳酸,也發現有大量的二羧酸和羥基酸。因此,盡早對臨床可疑患兒行血串聯質譜檢查及尿氣相質譜分析可為MADD患兒的早期診斷及治療提供幫助。
遲發型患兒可表現為肌病及肝功能異常,易出現漏診和誤診。本例患兒以轉氨酶升高就診,易誤診為肝臟相關性疾病,最終通過基因及腓腸肌活檢病理檢查以明確診斷。檢測到受檢者ETFDH基因的第13外顯子,c.1773_1774 del AT p.(Cys592※),該突變為無義突變,生物信息學軟件預測有致病可能,綜合考慮認為該突變為致病突變。c.389A>T p.(Asp130Val),該突變為錯義突變,生物信息學軟件預測有致病可能,綜合考慮認為該突變為可疑致病突變。結合受檢者父母樣本的檢測結果,這兩個突變分別位于不同的等位基因上。ETFDH基因突變引起的MADD晚發型主要臨床表現為脂質沉積性肌病,由于其對維生素B2治療敏感,故又稱核黃素反應性脂質沉積性肌病[5]。致病突變的攜帶者往往并不發展成為患者,故父母均為無癥狀攜帶者。攜帶致病突變的父母每次生育子女均有25%的可能為患者。
迄今為止MADD共報道了80余種致病突變,c.250G>A為我國南方MADD的致病性突變熱點[6],而本次檢測的兩個突變位點罕見報道,尤其c.1773_1774為新發現突變位點,且為致病突變。
MADD患兒在控制飲食同時補充核黃素、輔酶Q10和肉堿,可明顯改善癥狀及生化指標[7]。臨床上以轉氨酶升高為主伴有心肌酶升高、運動障礙者,應盡早進行分子遺傳學檢查,有條件者行腓腸肌肌肉活檢術,避免誤診的發生,早診斷、早治療對于遲發型MADD患兒至關重要,并可為患兒家庭提供準確的遺傳咨詢和產前診斷。
[1] 邢雅智.多種酰基輔酶A脫氫酶缺乏癥的診治進展[J].國際兒科學雜志,2010,37(5):518-521.
[2] Grünert SC. Clinical and genetical heterogeneity of late-onset multiple acyl-coenzyme A dehydrogenase deficiency[J]. Orphanet J Rare Dis,2014,9:117.
[3] Zhuo Z, Jin P, Li F,et al. A case of late-onset riboflavin responsive multiple acyl-CoA dehydrogenase deficiency (MADD) with a novel mutation in ETFDH gene[J]. J Neurol Sci,2015,353(1-2):84-86.
[4] Wen B, Li D, Li W, et al. Multiple acyl-CoA dehydrogenation deficiency as decreased acyl-carnitine profile in serum[J]. Neurol Sci,2015,36(6):853-859.
[5] 伏紅霞.核黃素反應性多種酰基輔酶A脫氫酶缺乏癥[J].中國醫師雜志,2015,17(9):1438-1440.
[6] Wang ZQ, Chen XJ, Murong SX, et al.Molecular analysis of 51 unrelated pedigrees with late-onset multiple acyl-CoA dehydrogenation deficiency(MADD) in southern China confirmed the most common ETFDH mutation and high carrier frequency of c.250G>A[J]. J Mol Med (Berl),2011,89(6):569-576.
[7] Cornelius N, Byron C, Hargreaves I, et al.Secondary coenzyme Q10 deficiency and oxidative stress in cultured fibroblasts from patients with riboflavin responsive multiple Acyl-CoA dehydrogenation deficiency[J]. Hum Mol Genet,2013,22(19):3819-3827.
Clinicalmanifestationandgeneanalysisofmultipleacyl-coadehydrogenasedeficiencywithtransaminaseincrease
TANYangfang,OUYANGWenxian,JIANGTao,TANGLian,SUYuxia,LIShuangjie.
DepartmentofHepatopathyCenter,HunanChildren'sHospital,Changsha410007,China
ObjectiveTo study the clinical manifestations of the patients with multiple acyl-coa dehydrogenase deficiency with transaminase increase, the laboratory tests, muscle biopsy and gene mutations, and to review the literature in order to provide the evidence for the early diagnosis and treatment of the disease.MethodsCollect the clinical data of a boy aged 7 years and 3 months, and take the blood samples of the child and his parents. The second-generation gene sequencing was used to detect the pathogenic gene, and gastrocnemius muscle biopsy was used to find out the muscle condition.ResultsThe electron microscopic results of the muscle biopsy showed that there was a lot of lipid droplets in the myofiber. The gene sequencing results showed that there were nonsense mutation of c.1773_1774 del AT p.(Cys592※) and missense mutation of c.389A>T p.(Asp130Val) in ETFDH genes of the child, which was considered to be complex heterozygous mutation. The parents were the carriers.ConclusionClinically there are people with transaminase increase complicated with increase of myocardial enzymes and movement disorders, in whom molecular genetic examinations should be performed as soon as possible. The patients with suitable conditions should be given gastrocnemius muscle biopsy, which can provide accurate hereditary consultation and prenatal diagnosis for the family of the patient.
Multiple acyl-coa dehydrogenase deficiency; Lipid storage myopathy; ETFDH gene; Child
R725
A
1674-3865(2017)05-0456-03
2017-05-23)
(本文編輯:吳迪)
410007 長沙,湖南省兒童醫院肝病中心
譚艷芳(1981-),女,醫學碩士,主治醫師。研究方向:小兒肝膽內科疾病的診治
李雙杰,E-mail:lesjie62@vip.sina.com
10.3969/j.issn.1674-3865.2017.05.031
