鋰離子電池電極材料表界面結構的原子尺度表征
2017-11-04 08:15:09仝毓昕張慶華
中國材料進展 2017年10期
仝毓昕,張慶華,谷 林
(中國科學院物理研究所,北京 100190)
特約專欄
2017-04-17
國家自然科學基金資助項目(51522212,51421002,51672307);科技部973計劃項目(2014CB921002)
仝毓昕,女,1996年生,碩士研究生
谷 林,男,1979年生,研究員,博士生導師,Email:l.gu@iphy.ac.cn
10.7502/j.issn.1674-3962.2017.10.04
鋰離子電池電極材料表界面結構的原子尺度表征
仝毓昕,張慶華,谷 林
(中國科學院物理研究所,北京 100190)
鋰離子電池充放電過程中,鋰離子的傳輸要穿過多種表面和界面,表界面的性質對電池的功率密度、能量密度、充放電效率、使用壽命、循環穩定性等具有重要的影響。表界面一般具有與體相不同的結構,在原子尺度上直接觀察不同電化學狀態下電極表界面的結構,有助于從更深層次認識電化學反應機理和性能演化規律,對于改善鋰離子電池性能具有重要的指導意義。闡述了球差校正透射電子顯微成像技術在研究電極材料表界面結構原子尺度研究中的應用,介紹了特殊的相界面、SEI、表面相變、表面摻雜等,探討了表界面原子尺度結構與性能的內在關聯,提出了改善電池性能的針對性建議,并對鋰離子電池未來的發展從提高能量密度、避免固液界面副反應和改善電池性能三個方面進行了展望。
鋰離子電池電極材料;球差校正掃描透射電子顯微鏡;表界面原子尺度結構;電化學反應機理;表面相變;相界面
1 前 言
鋰離子電池具有能量密度高、功率密度高、服役壽命長、無記憶效應等優點,已廣泛應用于消費電子、醫療電子、電動工具等小型可充放電電池領域[1,2],在混合動力汽車、航空航天、船舶艦艇等領域獲得了逐步推廣,并且在大規模智能電網儲能新興市場表現出巨大的應用前景[3]。隨著鋰離子電池應用的不斷擴大,對其性能的要求也在不斷提高,比如,更高的能量密度、更長的服役壽命、更低的成本、更好的安全性等,這就要求更加深入地認識鋰離子電池的微觀反應機制。
鋰離子電池通過鋰離子在正負極之間的遷移實現電能的儲存和轉化,在電子及離子的遷移過程中會涉及到多個尺度的表界面效應對電池性能產生影響。從宏觀尺度到微觀尺度,這些界面可以分為電極與電解液之間的固液界面、電極組成顆粒之間的固固界面、晶粒與晶粒之間的晶界、相與相之間的相界等。由于表界面處的原子排布、元素組成都與體相結構有較大差異,會導致材料在表界面處形成缺陷、化學反應活性位點或存在應力的積累,這些都會使材料表界面具有與體相結構完全不同的物理化學特性。同時,伴隨著鋰離子反復脫嵌的過程。鋰離子脫嵌和電子轉移,使得電極表面有可能形成新相,產生一些具有特殊結構的相界面。表界面結構影響電子和鋰離子的傳輸,從而影響電池的功率密度、能量密度、充放電效率和循環性能等。電化學循環中電極表界面的結構信息能夠促進更加深入的理解其化學反應機理和性能演化規律,從而優化和開發新電極材料。常用的材料結構表征手段有X射線衍射(XRD)、中子衍射(ND)、X射線吸收譜(XAS)、電子顯微術等。通過XRD、ND、XAS能夠得到原子的占位、組成元素的價態和周圍的化學環境等信息,但是由于空間分辨率不足,只能反映材料結構的整體特征[4-6]。
電子顯微術包括透射電子顯微術(TEM)、掃描電子顯微術(SEM)和掃描透射電子顯微術(STEM)等,隨著球差校正技術的出現,透射電子顯微鏡的分辨率已經能達到亞埃量級[7,8],高角度環形暗場掃描透射電子顯微技術(HAADF-STEM)可得到原子分辨像[9],但成像襯度強烈依賴于原子序數Z,不能直接觀測輕原子,比如電池電極材料中和重要的鋰和氧。球差校正環形明場掃描透射電子顯微成像技術(ABF-STEM)不僅可以得到能夠直接解釋的高分辨電子顯微像,而且圖像襯度與原子序數Z的1/3次方成正比,實現了輕重原子同時成像[10]。目前,采用該方法已經實現了對鋰、硼、碳、氮、氧甚至氫的直接成像[10-13],并在鋰離子電池電極材料顯微結構的表征中得到了廣泛應用。本文綜述了球差校正掃描透射電子顯微鏡在研究幾種典型電極的表界面結構中的應用,并探討了結構對電池性能的具體影響。其中,常見的正極材料包括橄欖石型LiFePO4、層狀LiCoO2和Li2MnO3、尖晶石型LiMn2O4和硫等,負極材料包括層狀石墨類材料和尖晶石型Li4Ti5O12等。
2 鋰離子電池電極材料的表界面結構
2.1 正極材料的表界面結構
2.1.1 橄欖石型LixFePO4中的相界面——“二階”結構
在1997年Goodenough等[14]首次提出LiFePO4可以作為鋰離子電池的正極材料,雖然LixFePO4的發現時間早,已經進行了很多研究,在實際運用中取得了一些成果,但是仍然存在許多問題沒有解決,其中,關于電極脫嵌鋰過程中LiFePO4到FePO4的相轉變的微觀反應機制一直尚未明確。研究發現LiFePO4的充放電過程中存在明顯的電位平臺,認為 LiFePO4在脫嵌鋰過程中發生兩相分離反應,即:LiFePO4?FePO4+Li++e-。為了弄清相轉變的微觀反應機制,提出了很多模型,包括核-殼模型(core-shell model)[14]、馬賽克模型(mosaic model)[15]、新核-殼模型(new core-shell model)[16]、多米諾模型(domino-cascade)[17],但是都沒能對兩相結構界面的細節進行描述。
谷林等[18]采用球差校正環形明場成像技術,直接觀察到了部分脫鋰的LiFePO4單晶納米線(d=65 nm)中鋰離子隔行脫出的現象,如圖1所示,其中黃色圈和橘色圈分別表示鋰原子存在位置和脫出位置。與石墨插層化合物中出現的單相“二階”結構類似,與之前所提出的兩相反應的模型均不相同。之后,Malik等[19]通過Monte Carlo模擬計算也發現脫嵌鋰過程中存在單相反應,形成類似階結構的亞穩中間相,從另一角度解釋了低電子和離子電導的LiFePO4可以快速充放電的原因。進一步研究發現,“階”結構主要存在于LiFePO4和FePO4兩相之間,形成LiFePO4/“階”界面相/FePO4的三相共存結構,而且“階”結構界面存在明顯的尺寸效應[20,21],即當顆粒尺寸由100 nm減小到70 nm時,界面區的“階”結構由2 nm增加到15 nm。另外,索鎏敏等[21]研究了部分脫鋰的Nb摻雜的LiFePO4(d= 200 nm)納米顆粒的原子結構,觀察到了LiFePO4和FePO4兩相界面處的高度有序的“階”結構,表明Nb的摻雜并不會影響“階”結構的形成,說明了形成的單相“階”結構是本征的亞穩相或者中間相。孫洋等[22]通過密度泛函理論研究“階”結構的形成機制,發現兩相反應在熱力學上是有利的,熱力學和動力學影響因素相互競爭導致LiFePO4在脫鋰過程中出現LiFePO4/“階”界面相/FePO4的三相共存結構。
另外,當電解質中存在水汽和羥基時,在LiFePO4表面會生成惰性的LiFePO4(OH),這使電池循環性能差出現容量衰減,因此提高電極材料表面結構的穩定性對改善電池性能起著很關鍵的作用[23]。
LiFePO4作為鋰離子電池的正極時,其電導率低是影響其性能的重要因素。為改善其電化學性能,提出了碳包覆納米LiFePO4顆粒,碳包覆層不僅可以改善納米顆粒電極材料表面間的電子轉移,而且為電子在納米顆粒電極材料之間和納米顆粒電極材料和集流體之間的電子傳導提供了額外的途徑,明顯改善了電池的倍率性能。
2.1.2 層狀LiCoO2的表面結構相變
LiCoO2是第一代商業化鋰離子電池的正極材料,并且現在仍然主導著便攜式電子設備的市場,LiCoO2具有多種結構,包括O1型、O2型和熱力學穩定的O3型,其中O1型中的氧是ABAB型排列,O2型中的氧是ABAC排列,O3型中氧是ABCABC排列[24-26]。盧俠等[27]采用球差校正的掃描透射電子顯微鏡成像技術首次O3型LiCoO2電化學過程中直接觀察到了O2型結構,LiCoO2經過第一個脫嵌鋰過程后,表面位置的Co原子從過渡金屬層轉移到Li層,產生Li-Co反位缺陷,使得表面Li層的襯度相比于體相明顯增加,如圖2所示,材料的表面結構從層狀結構變成了鹽巖結構。表面Co原子占據Li層的八面體位置會導致表面層間距與體相不同,阻礙鋰離子在Li層的遷移,從而影響電池的倍率性能。 Zheng等[28]對LiNi0.8Co0.15Al0.05O2三元正極材料的研究表明,第一次電化學循環后,電極表面過渡金屬離子遷移至Li層,導致表面結構相變,并認為陽離子互占位使表面結構相變是電化學循環可逆容量損失的主要原因。為了提高其可逆容量,可以考慮采用表面包覆的方法,來抑制電化學循環過程中結構相變。此外,可以采用摻雜的方法,使更多的鋰離子從晶格中脫出,提高電池的能量密度。降低LiCoO2的成本和提高較高溫度下的循環性能也是重要的研究方向。

圖1 不同充電狀態下LiFePO4沿[010]方向的ABF像:(a)初始狀態,(b)完全充電狀態,(c)半充電狀態。圖中黃色圈和橘色圈分別表示鋰占據和脫出的位置[18]Fig.1 ABF micrographs taken along [010] zone axis showing Li ions of partially delithiated LiFePO4 at every other row: (a) pristine material with the atomic structure of LiFePO4 shown as inset, (b) fully charged state with the atomic structure of FePO4 shown for comparison, and (c) half charged state showing the Li staging. Note that Li sites are marked by yellow circles and the delithiated sites are marked by orange circles[18]

圖2 LiCoO2納米顆粒充放電前后沿[010]方向的HAADF像和ABF像:(a)和(b)初始狀態; (c)和(d)放電至3.0 V[27]Fig.2 HAADF and ABF images taken along [010] of LiCoO2 nanoparticles after and before charging: (a) and (b) pristine LiCoO2, (c) and (d) discharge to 3.0 V[27]
2.1.3 層狀Li2MnO3中的表面摻雜改性
2001年,Thackeray和Johnson[29]提出了一種具有復合結構的材料,其結構是一層Li2MnO3和一層LiMO2或一層LiM2O4(M=Fe,Mn,Ni,Co,etc.)復合而成,并且證明了其比容量達到250~300 mAh/g[29,30],使用該材料作為正極的鋰離子電池的能量密度達到了300 WhKg-1,比LiCoO2作為正極材料時最大能量密度220 WhKg-1大很多。富鋰層狀氧化物作為鋰離子電池的正極材料,具有比容量高、成本較低、安全性好的優點,近年來受到廣泛關注。但是,富鋰層狀材料作為鋰離子電池正極時,存在循環過程中容量損失和電壓衰減的問題[31],這些問題與鋰離子的擴散系數低有很大關系。
有研究表明采用Al2O3、AlPO4[32]、AlF3[33]進行表面包覆可以在一定程度上緩解這些問題,因為表面包覆可以抑制SEI[34]和表面相[35]的形成,SEI和表面相均是導致鋰離子擴散系數低的原因[36,37]。但是這種方法只能提高電極表面區鋰離子的擴散系數,而不能提高電極內部。此外,納米結構使電子和鋰離子的擴散路徑顯著縮短以改善電池的倍率性能,但是同時也會導致體積膨脹和副反應等問題。
Qing等[39]提出電極表面Na+濃度梯度摻雜的方法,能夠提高富鋰層狀顆粒電極材料的擴散動力學,從而改善電池的性能。利用Na+濃度驅動擴散,通過煅燒在熔融的NaCl中的富鋰層狀材料實現表面Na+濃度梯度摻雜,電極材料結構示意圖如圖3。采用球差校正掃描透射電子顯微鏡觀察電極表面,如圖4,表明電極表面Na+濃度梯度摻雜后Na+進入到Li層。由于填塞效應使得存在很多缺陷的富鋰層狀結構更加穩定,而且使(003)和(104)晶面間距增大促進了Li+的擴散,如圖5。純凈的富鋰材料和表面Na+濃度梯度摻雜富鋰材料的充放電曲線如圖6,可以看出表面Na+濃度梯度摻雜富鋰材料具有更大的可逆容量和更好的循環性能。通過對電極材料表面近平衡結構的觀察,深入了解電化學循環中的脫嵌鋰機制,進而針對性地指導電極材料的改性。摻雜改變鋰離子擴散的動力學,為改善電極材料中的鋰離子擴散和表面結構穩定性提供了思路。
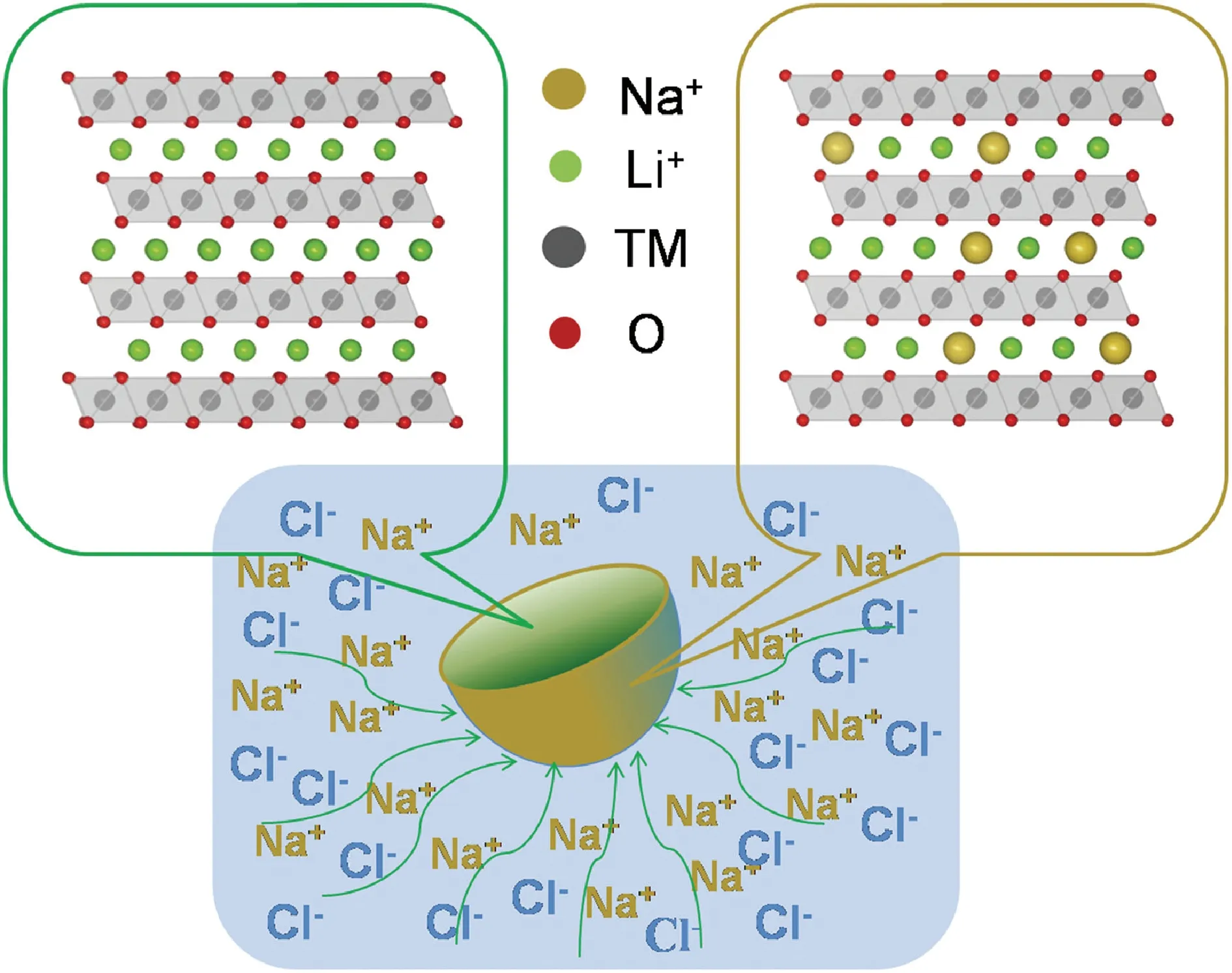
圖3 表面Na+濃度梯度摻雜電極結構示意圖[39]Fig.3 Schematic illustration of the structure design of gradient surface Na+ doping Li-rich material[39]

圖4 表面Na+濃度梯度摻雜的富鋰材料表面的沿[010]方向的STEM照片:(a)HAADF圖,(b)ABF圖和(c)ABF的襯度曲線[39]Fig.4 STEM of gradient surface Na+ doped Li-rich material surface along [010] zone axis: (a) HAADF image, (b) ABF image, and (c) ABF line profile[39]
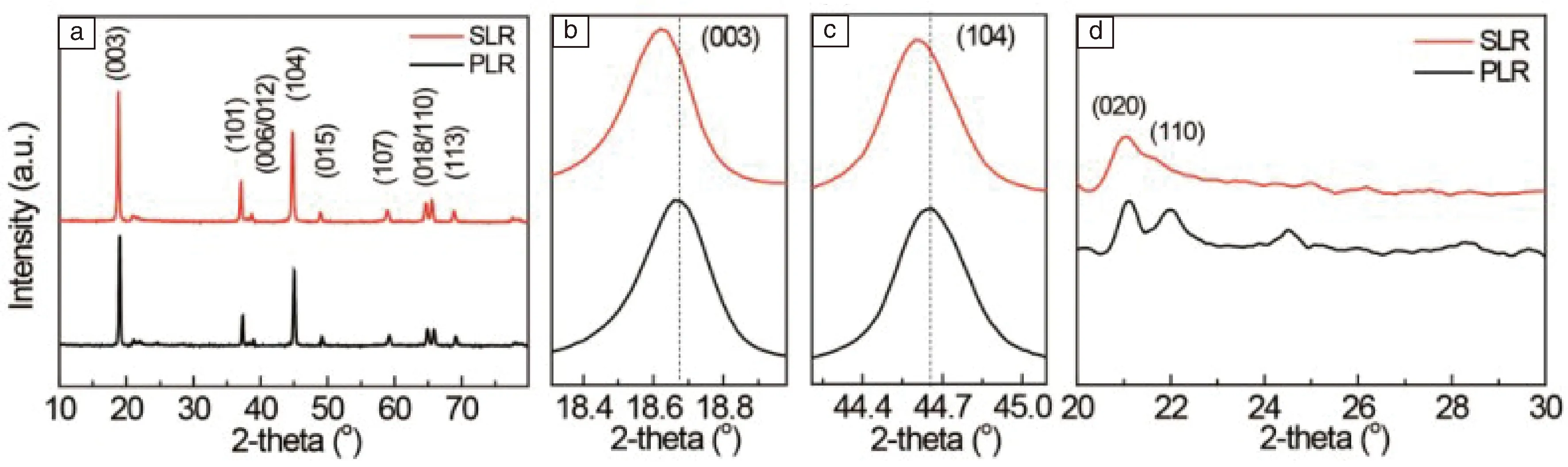
圖5 (a)~(d) 純凈的富鋰材料和表面Na+ 濃度梯度摻雜富鋰材料的XRD譜圖Fig.5 XRD pattern of pristine Li-rich material and gradient surface Na+ doped Li-rich material

圖6 純凈的富鋰材料和表面Na+ 濃度梯度摻雜富鋰材料在0.1 C時的充放電曲線(a)和(b);純凈的富鋰材料和表面Na+ 濃度梯度摻雜富鋰材料在0.2 C時的倍率性能和循環穩定性(c)和(d);所有曲線均是在25 ℃時電壓在2.0~4.7 V范圍下得到的[34]Fig.6 Charge-discharge curves of pristine Li-rich material and gradient surface Na+ doped Li-rich material at 0.1 C, respectively (a) and (b). Rate performance (c) and cycling stability (d) tested at 0.2 C of pristine Li-rich material and gradient surface Na+ doped Li-rich material. All of them were tested at 25 ℃ with voltage ranging from 2.0 to 4.7 V versus Li+/Li[34]
2.1.4 尖晶石型LiMn2O4的表面結構相變
尖晶石型LiMn2O4是一種重要鋰離子電池的正極材料,具有功率密度高、成本低、安全性能好等優點,并且已經成功地應用于電動汽車[40]。但是,LiMn2O4在電化學循環和長時間存儲過程中會出現嚴重的容量衰減的問題,尤其是在溫度升高的情況下。對于出現該問題的原因提出了很多理論,包括在高電位下電極材料與電解液發生反應[41]、尖晶石結構到巖鹽結構的轉變導致非平衡鋰化[42]和LiMn2O4表面錳溶解[43,44]等。其中,LiMn2O4表面錳溶解被認為是電池容量衰退的主要原因,但是其具體機理還待進一步研究,而且錳溶解的速率會隨著LiMn2O4中鋰含量的減小而增大,其原因也尚不明確。
唐代春等[45]采用球差校正掃描透射電子顯微鏡高角度環形暗場成像技術觀察了不同狀態下LiMn2O4的表面結構,如圖7,脫鋰的LiMn2O4的表面Mn原子從16d位置遷移至Li原來所占據的8a位置,生成了Mn3O4相,充電電壓升高,表面相變區域越大。放電過程中,表面中的Mn3O4相逐漸減少,當放電到3.0 V時,Mn3O4相基本消失,表面相與體相結構完全相同,充放電過程中電極表面的相變示意圖如圖8。電極表面Mn3O4相在充放電狀態下的出現和消失被認為是錳溶解的主要原因,并且提出了錳溶解的兩種可能的機制:①Mn3O4中四面體位置的Mn2+溶于電解液后形成與電極內部相似的結構;②Mn3O4完全溶解于電解液中。由此可見,表面結構相變導致錳溶解,損耗電極活性物質造成容量衰減,循環性能差,要改善電池循環性能就要穩定電極表面結構,可以考慮采用表面包覆或者形成SEI膜的方法。

圖7 不同充放電狀態下LiMn2O4 沿(110)帶軸的HAADF像:(a)充電至4.1 V;(b)充電至4.3 V;(c)放電至4.0 V;(d)放電至3.0 V。在所選擇區域的放大圖像中,位于16d和8a位置的Mn原子列襯度分別用藍色和橙色球表示,表面相變區域與體相的邊界由綠色虛線標示[45]Fig.7 HAADF images taken along the (110) zone axis of the LiMn2O4 spinel cathode charged to 4.1 V (a) and 4.3 V (b) and discharged to 4.0 V (c) and 3.0 V (d). Magnified views of selected regions are shown in the right panels, where the contrast corresponding to the Mn columns at 16d and 8a sites is indicated by blue and orange spheres, respectively. The boundary between the bulk and the surface regions is marked by the green dashed line [45]

圖8 充放電的LiMn2O4顆粒的相變示意圖。綠色的透明度代表了LixMn2O4中鋰的濃度,其中完全透明的綠色代表λ-MnO2 相,完全不透明的綠色代表LiMn2O4相,橙色代表Mn3O4相。充電過程中,Mn3O4相在表面區形成,并在電壓為4.3 V時達到最大,之后的放電過程中Mn3O4相逐漸減少,在放電結束時幾乎全部消失[45]Fig.8 Schematic of phase evolution of a LiMn2O4 particle upon charge/discharge. The lithium concentration of LixMn2O4 is represented by the transparency of green color where the fully transparent green color indicates λ-MnO2 and the fully opaque green color indicates LiMn2O4. The Mn3O4 phase is indicated by the orange color. During the charge process, the Mn3O4 phase forms at the surface region and its amount reaches a maximum after being charged to 4.3 V. The surface Mn3O4 phase decreases gradually on the subsequent discharge process, and it is almost negligible at the end of discharge[45]
2.1.5 鋰硫二次電池中硫電極的SEI膜和相界面
鋰硫二次電池具有很高的理論比容量(1673 mAhg-1)和能量密度(2600 WhKg-1)[46],并且硫的成本低,儲量非常豐富。鋰硫電池已經成為電池領域的研究熱門,但是仍然存在一些問題有待解決,硫是絕緣體,電極在充放電過程中發生膨脹,鋰硫之間化學反應復雜以及多硫化物的溶解造成的穿梭效應等都造成了鋰硫電池性能不理想。為了提高硫的導電性,常采用良好導電性的材料與其復合,其中碳材料是使用最多也最有效的載體,比如乙炔黑、多孔碳、石墨烯、碳納米管等。在液體電解質的鋰硫電池中,由于穿梭效應造成容量衰減和過充現象,為了解決該問題嘗試了電極添加劑、電解質添加劑、設計阻擋層、采用固體電解質等方法。常見的電解質添加劑有LiNO3[47], LiBOB[48]和P2S5[49]等。LiNO3作為電解液添加劑對提高鋰硫電池庫侖效率效果尤為明顯, 目前被廣泛使用。 Aurbach等[47]通過紅外光譜(FTIR)及光電子能譜(XPS)等譜學手段對加入LiNO3的鋰硫電池進行了研究,認為LiNO3的加入有助于在金屬鋰負極表面原位形成一層致密且穩定的SEI膜, 從而有效阻擋溶解于電解液中多硫離子進一步與金屬鋰反應。
要改善鋰硫電池的性能,先要清楚其電池電化學反應的微觀機制。采用原位X射線衍射方法研究其反應機制,但是空間分辨率不高,不能表征反應的細節。楊振中等[50]采用原位透射電子顯微鏡并結合電子能量損失譜(EELS)研究了碳包覆的硫納米顆粒的鋰化過程,得到了鋰硫電池的電化學反應機理。進行固態鋰硫電池電極反應過程的示意圖如圖9,鋰化過程中,硫部分轉化為多晶硫,部分轉化為Li2S,發生了相分離,這與之前報道的存在電壓平臺一致[51]。相分離不僅減小了擴散距離,而且S/Li2S相界面的網狀結構有助于電子和離子的傳導。隨著鋰化進行,多晶硫全部轉化為Li2S。雖然實驗中碳包覆硫電極很穩定,但是仍與實際情況有一定差別,高能電子束和真空都有可能改變相變動力學,盡管如此,實驗中對硫電極的反應機制的探索對固態鋰硫電池性能的改善提供了新的思路。
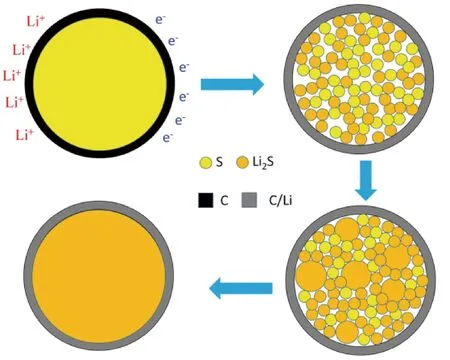
圖9 固態鋰硫電池的電化學反應機制示意圖:(a)碳包覆的純凈的硫樣品;(b)鋰化初始階段S和Li2S相分離;(c)S相減少,Li2S相增多;(d)完全鋰化后純的Li2S相[50]Fig.9 Schematic diagram of the electrochemical mechanism of a solid-state Li-S battery: (a) Pristine S sample with the carbon coating; (b) The phase separation of S and Li2S during the early stage of the lithiation process; (c) The phase of S reduces and that of Li2S increases as the lithiation process progresses; (d) The pure Li2S phase formed at the end of lithiation process[50]
要改善鋰硫二次電池的性能,一方面可以考慮設計比表面積大的硫載體,充分利用硫,提高能量密度,另一方面,抑制穿梭效應,形成SEI或者采用固體電解質等。雖然采用固體電解質避免了穿梭效應,但是固態電解質的電導率低,因此,要開發新的固體電解質或電極活性物質,減小界面接觸電阻。
2.2 負極材料尖晶石型Li4Ti5O12的相界面結構
尖晶石型Li4Ti5O12作為一種重要的鋰離子電池的負極材料,盡管其成本較高,導電性差,但是由于其循環壽命長,較高的嵌鋰電位,“零應變”等優點,仍然在鋰離子動力電池領域展現出巨大的應用前景[52]。但是一個較嚴重的問題是Li4Ti5O12在充放電以及儲存過程中,電極會發生脹氣,影響電池性能[53]。
從晶體學結構看,尖晶石型Li4Ti5O12屬于立方晶系,它可以看做Li[Li1/3Ti5/3]O4,其中Li, [Li1/3Ti5/3]和O分別占據8a,16d和32e位置。在電化學循環過程中,8a位置處的Li和嵌入的Li進入到空的16c位置,直到所有的16c位置被填滿,這將導致尖晶石型Li4Ti5O12轉變成巖鹽結構的Li7Ti5O12。Li4Ti5O12的充放電過程中表現出一非常平坦的電壓平臺,表明鋰離子在Li4Ti5O12晶格中的脫嵌是一個兩相反應[54],即Li4Ti5O12+Li++e-→Li7Ti5O12。采用球差校正的環形明場成像技術,盧俠等[55]首次在原子尺度上直接觀察到了部分鋰化的納米顆粒中Li4Ti5O12和Li7Ti5O12的兩相共存結構,如圖10所示,進而證實了兩相反應機制。Li4Ti5O12(區域1)和Li7Ti5O12(區域2)兩相形成完全共格界面,如圖10a和10b中的黃線所示,使其具有零應變的特性。如圖10d中的黑點所示,Li7Ti5O12由表面向內部延伸,在兩相界面處Li7Ti5O12中的鋰離子偏離16c的位置,靠近鄰近的O原子,與第一性原理計算的結果一致,如圖10e。
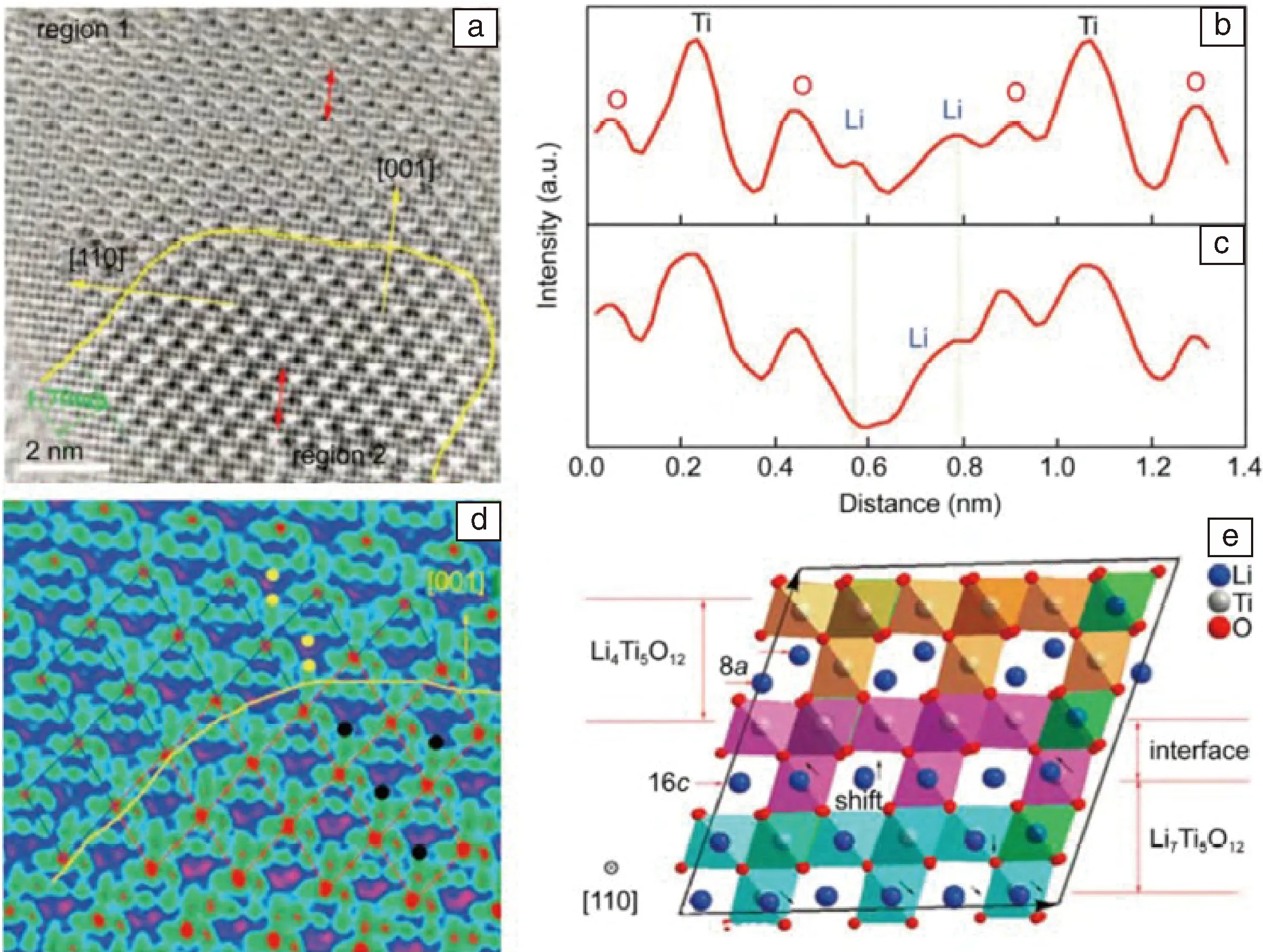
圖10 Li4Ti5O12/Li7Ti5O12兩相共存區沿[110]方向的ABF像:(a)黃色線表示兩相界面, 綠色平行線標示不同于體相的表層結構;(b) Li4Ti5O12與(c) Li7Ti5O12的襯度曲線;(d)放大的兩相邊界結構ABF像;(e)第一性原理模擬的Li4Ti5O12/Li7Ti5O12兩相界面結構示意圖[54]Fig.10 Interfacial structure in partially lithiated Li4Ti5O12 sample viewed along the [110] direction: (a) ABF image near the interface between Li4Ti5O12 phase (region 1) and Li7Ti5O12 phase (region 2). The yellow line indicates the boundary of the interface; (b) and (c) Line profiles of region 1 and region 2, respectively; (d) Colored ABF image of the two phases near the interface, where the 8a sites occupied in Li4Ti5O12 and the 16c sites occupied in Li4Ti5O12 are marked as yellow and black dots, respectively; (e) The relaxed interfacial structure simulated by DFT calculations, where the Li 16d-O octahedrons are shown in green. The remarkable shifts of Li+ ion at the 16c sites in Li7Ti5O12 region are indicated by black arrows [54]
為了解決Li4Ti5O12的導電性差,鋰離子擴散系數低的問題,將其納米化并在表面進行包覆被認為是有效方法[56-58]。王永慶等[59]控制水熱反應中的Li/Ti投料比和前驅體的煅燒溫度,首次合成了側面包覆金紅石TiO2的Li4Ti5O12納米片,如圖11所示。電化學測試發現金紅石TiO2包覆的Li4Ti5O12表現出更加優異的動力學特性,表明控制電極表面結構可以有效地改善電池的電化學性能,表面修飾為改善電極材料的性能提供了新的思路。
之后,盧俠等[60]采用球差校正掃描透射電子顯微成像技術,研究不同化學狀態下球形Li4Ti5O12納米顆粒的表面結構,如圖12所示,發現表面(110)面上dAB和dCD的長度是體相的1.15倍,表面(111)面上dAB和dCD的長度是體相的1.5倍,表明電極表面發生了結構弛豫,甚至表面重構。表面弛豫導致16d位置處的Ti原子遷移至Li離子晶格中的16c位置,在電極表面產生了一些與內部不同的Ti-O配位體。除了表面弛豫和重構,表面懸鍵也是重要的電化學活性中心,從圖12中可以看出電極表面有很多Ti原子,可能是不飽和的Ti-O鍵,當電極暴露在空氣中時,將會吸附水汽和CO2。因此,這種類型的Ti原子(或者Ti-O)很容易形成Ti-O:R(R: Li+、H+、CH+或其他有機原子團),這會破壞TiO6八面體的對稱性,造成Ti-O鍵不可逆地伸長/縮短并且形成了電子/空穴捕獲中心。表面結構弛豫和重構,表面懸鍵這些微小的表面結構變化都可能在脫嵌鋰過程中發生,但是具體的分子反應機制和釋放氣體的原因,還待進一步闡明。
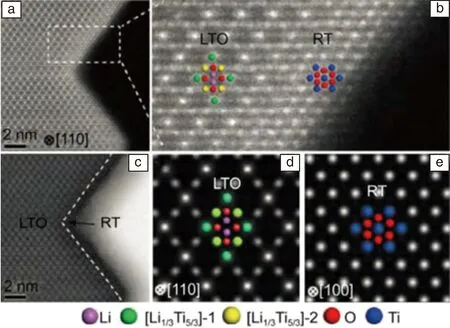
圖11 金紅石 TiO2 包覆的Li4Ti5O12 納米片 STEM 照片:(a)和(b)納米片沿[110]晶向的HAADF像; (c)對應的ABF像;(d)模擬的Li4Ti5O12和(e)金紅石TiO2的HAADF像[59]Fig.11 STEM images of LTO-RT-600: (a) and (b) HAADF as well as (c)corresponding ABF images of LTO-RT-600 NSs. The simulated HAADF images of (d) LTO projected from [110] direction and (e) rutile-TiO2 projected from [100] direction. The insets show the arrangements of atoms[59]

圖12 Li4Ti5O12原子尺度表面結構的HAADF像:(a) 沿[[60]Fig.12 Images of atomic-scale surface structures of Li4Ti5O12 samples obtained with STEM-HAADF techniques:(a) Clean [110] surface; (b) [110] surface with obvious Li-Ti disorders in the 8a site; (c) [111] and (d) [121] surfaces[60]
Li4Ti5O12的“零應變”特性有利于電池和電極材料的穩定,可以實現長的循環壽命,考慮到其脹氣問題可能是由于電極表面活性高造成的,因此,可以采用表面修飾的方法來降低其表面反應活性。
3 結 語
鋰離子電池的表界面問題是影響鋰離子電池性能最關鍵的問題之一,也是鋰離子電池領域最活躍的研究熱點之一。對表界面問題的深入理解需要依賴多尺度、高精度的分析手段。球差校正透射電子顯微鏡以及原位透射電鏡的發展,使得對電極材料的表面界面結構進行原子尺度的原位表征變為可能,極大地促進了人們對電化學反應過程中表界面反應機制的深入理解,對電極材料的設計和開發具有指導意義。
目前,發展高能量密度液態鋰離子電池仍然是科研界及產業界關注的重點。傳統的表征手段已經無法滿足人們對深層次物理化學問題理解的需要。球差電子顯微鏡的發展,讓研究人員能夠從原子尺度理解材料在電化學反應過程中經歷的種種變化,曾經困擾著人們的許多難以解決的科學技術難題,如:材料表面相變規律對材料電化學循環穩定性的影響、材料內部相的分布與遷移規律對材料動力學性質的改變、相變過程對材料產氣及電壓衰減的影響等,都在逐漸為人們所理解。以這些理解為基礎建立的材料設計方案已經初見成效,正推動著液態鋰離子電池能量密度向著理論極限不斷邁進。
未來,固態化必將是鋰離子電池以及所有液態電池發展的終極目標。固態電池以其優異的能量密度以及安全特性正逐漸為人們所關注,而固態化帶來的新的挑戰也等待著人們去探索。電極與電解質的固固界面、全固態條件下材料新的表界面相變機理以及固相間互擴散形成的過渡相等等,這些問題都制約著固態電池的發展。球差校正透射電子顯微鏡以及原位透射電鏡技術在解決這些關鍵問題上具有極大的優勢,能夠幫助人們快速精確地找到問題背后最本征的物理化學起因,從而對癥下藥快速解決問題。希望在球差電子顯微鏡以及其他高精度表征手段的發展與幫助下,能夠帶領我們在新的領域不斷進取,解決關鍵的基礎科學、技術難題。
References
[1] Tarascon J M, Armand M.Nature[J], 2001, 414(6861): 359-367.
[2] Thackeray M M, Wolverton C, Iasscs E D,etal.EnergyEnvironSci[J], 2012, 5(7): 7854-7863.
[3] Dunn B, Kamath H, Tarascon J M,etal.Science[J], 2011, 334(6058): 928-935.
[4] Reimers J N, Dahn J R.JElectrochemSoc[J], 1992, 139(8): 2091-2097.
[5] Sharma N, Peterson V K, Elcombe M M,etal.JPowerSources[J], 2010, 195(24): 8258-8266.
[6] Yoon W S, Grey C P, Balasubramanian M,etal.ChemMater[J], 2003, 15(16): 3161-3169.
[7] Haider M, Uhlemann S, Schwan S,etal.Nature[J], 1998, 392(6678): 768-769.
[8] Batson P E, Dellby N, Krivanek O L.Nature[J], 2002, 418(6898): 617-620.
[9] Pennycook S J, Jesson D E.PhysRevLett[J], 1990, 64(8): 938-941.
[10] Findlay S D, Shibata N, Sawada H,etal.Ultramicroscopy[J], 2010, 110(7): 903-923.
[11] Lin Y R, Ho C Y, Hsieh C Y,etal.ApplPhysLett[J], 2014, 104(12): 121909.
[12] Gu L, Zhu C B, Li H,etal.JAmChemSoc[J], 2011, 133(13): 4661-4663.
[13] Ishikawa R, Okunishi E, Sawada H,etal.NatMater[J], 2011, 10(4): 278-281.
[14] Padhi A K, Nanjundaswamy K S, Goodenough J B,etal.JElectrochemSoc[J], 1997, 144(4): 1188-1194.
[15] Anderson A S, Thomas J O.JPowerSources[J], 2001, 97(3): 498-502.
[16] Laffont L, Delacout C, Gibt P,etal.ChemMater[J], 2006, 18(23): 5520-5529.
[17] Delmas C, Maccario M, Croguennec L,etal.NatMater[J], 2008, 7(8): 665-671.
[18] Gu L, Zhu C B, Li H,etal.JAmChemSoc[J], 2011, 133(13): 4661-4663.
[19] Malik R, Zhou F, Ceder G.NatMater[J], 2011, 10(8): 587-590.
[20] Zhu C B, Gu L, Suo L M,etal.AdvFunctMater[J], 2014, 24(3): 312-318.
[21] Suo L M, Han W Z, Lu X,etal.PhysChemChemPhys[J], 2012, 14(16): 5363-5367.
[22] Sun Y, Lu X, Xiao R J,etal.ChemMater[J], 2012, 24(24): 4693-4703.
[23] Cuisinier M, Martin J F, Dupre N,etal.ElectrochemCommun[J], 2010, 12(2): 238-241.
[24] Mizushima K, Jones P C, Wiseman P J,etal.MaterResBull[J], 1980, 15(6): 783-789.
[25] Delmas C, Braconnier J J, Hagenmuller P.MaterResBull[J], 1982, 17(1): 117-123.
[26] Carlier D, Saadoune I, Suard E,etal.SolidStateIonics[J], 2001, 144(3): 263-276.
[27] Lu X, Sun Y, Jian Z L,etal.NanoLett[J], 2012, 12(12): 6192-6197.
[28] Zheng S J, Huang R, Makimura Y,etal.JElectrochemSoc[J], 2011, 158(4): A357-A362.
[29] Thackeray M M, Johnson C S, Vaughey J T,etal.MaterChem[J], 2005, 15(23): 2257-2267.
[30] Thackeray M M, Kang S H, Johnson C S,etal.JMaterChem[J], 2007, 17(30): 3112 -3125.
[31] Verde M G, Liu H D, Carroll K J,etal.ACSApplMaterInterfaces[J], 2014, 6(21): 18868-18877.
[32] Cho J, Kim T G, Lee J G,etal.PowerSources[J], 2005, 146(1): 58-64.
[33] Rosina K J, Jiang M, Zeng D,etal.MaterChem[J], 2012, 22(38): 20602-20610.
[34] Jiang M, Key B, Meng Y S,etal.ChemMater[J], 2009, 21(13): 2733-2745.
[35] Xu Bo, Fell R C, Chi M,etal.EnergyEnvironSci[J], 2011, 4(6): 2223-2233.
[36] Yu X Q, Liu Y C, Gu Lin,etal.AdvEnergyMater[J], 2014, 4(5): 1300950.
[37] Wu F, Li N, Su Y F,etal.JMaterChem[J], 2012, 22(4): 1489-1497.
[38] Wei G Z, Lu X, Ke F S,etal.AdvMater[J], 2010, 22(39): 4364-4367.
[39] Qing R P, Shi J L, Xiao D D,etal.AdvEnergyMater[J], 2016, 6(6): 1501914.
[40] Park O K, Cho Y, Lee S,etal.EnergyEnvironSci[J], 2011, 4(5): 1621-1633.
[41] Pistoia G, Antonini A, Rosati R,etal.ElectrochimActa[J], 1996, 41(17): 2683-2689.
[42] Thackeray M M, Shao-Horn Y, Kahaian A J,etal.ElectrochemSolid-StateLett[J], 1998, 1(1): 7- 9.
[43] Jang D H, Shin Y J, Oh S M,etal.ElectrochemSoc[J], 1996, 143(7): 2204-2211.
[44] Zhan Chun, Lu Jun, Kropf A J,etal.NatCommun[J], 2013, 4(9): 2437.
[45] Tang D C, Yang S, Yang Z Z,etal.ChemMater[J], 2014, 26(11): 3535-3543.
[46] Bruce P G, Freunberger S A, Hardwick L J,etal.NatMater[J], 2012, 11 (1): 19-29.
[47] Aurbach D, Pollak E, Elazari R,etal.JElectrochemSoc[J], 2009, 156(8): A694-A702.
[48] Xiong S Z, Kai X, Hong X B,etal.Ionics[J], 2012, 18(3): 249-254.
[49] Lin Z, Liu Z C, Fu W J,etal.AdvFunctMater[J], 2013, 23(8): 1064-1069.
[50] Yang Z Z, Zhu Z Y, Ma J,etal.AdvEnergyMater[J], 2016, 6(20): 1600806.
[51] Nagao M, Imade H, Narisawa,etal.JPowerSources[J], 2013, 222(2): 237-242.
[52] Zhu G N, Wang Y G, Xia Y Y,etal.EnergyEnvironSci[J], 2012, 5(5): 6652-6667.
[53] He Y B, Li B H, Liu M,etal.SciRep[J], 2012, 2(12): 913.
[54] Ohzuku T, Ueda A, Yamamoto N J,etal.ElectrochemSoc[J], 1995, 142(5): 1431-1435.
[55] Lu X, Zhao L, He X Q,etal.AdvMater[J], 2012, 24(24): 3233-3238.
[56] Chen L, Li X L, Liu H J,etal.JElectrochemSoc[J], 2007, 154(7): A692-A697.
[57] Park K S, Benayad A, Kang D J,etal.JAmChemSoc[J], 2008, 130(45): 14930-14931.
[58] Zhao L, Hu Y S, Li H,etal.AdvMater[J], 2011, 23(11): 1385-1388.
[59] Wang Y Q, Gu L, Guo Y G,etal.JAmChemSoc[J], 2012, 134(18): 7874-7879.
[60] Lu X, Gu L, Hu Y S,etal.JAmChemSoc[J], 2015, 137(4): 1581-1586.
The Atomic-Scale Characterization of Surfaces and Interfaces of Electrode Materials for Lithium-ion Batteries
TONG Yuxin, ZHANG Qinghua, GU Lin
(Institute of Physics, Chinese Academy of Sciences, Beijing 100089, China)
Lithium-ion is transferred through a variety of surfaces and interfaces in Lithium ion batteries during the process of charging and discharging. The properties of surface and interface of electrode have great influences on the power density, energy density, the rate performance, service life, as well as cycling stability. In general, the structure of surfaces and interfaces of materials are different from that of bulk, direct observation upon atomic-scale structure of electrode in different electrochemical state contributes to studying electrochemical reaction mechanism and evolution of properties and is of instructive significance to improve the properties of lithium-ion batteries. In this paper, we review the recent progress in investigations of surfaces and interfaces structure of electrode materials, introduce special interface, SEI, the phase transition of surface and doping of surface, discuss the inherent correlation between the atomic-scale structures of surfaces and interfaces of electrode materials and their performance, raise some advices to improve the performance of lithium-ion batteries and look forward to the development of lithium-ion batteries in the aspect of increasing energy density, averting side reaction of solid electrode and liquid electrolyte as well as properties improvement.
lithium-ion batteries; aberration-corrected scanning transmission electron microscopy; atomic scale surface and interface structure; electrochemical reaction mechanism; the phase transition of surface; interface
O646.21
A
1674-3962(2017)10-0708-10
(編輯 吳 琛)
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
當代陜西(2020年13期)2020-08-24 08:22:02
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
制造技術與機床(2017年5期)2018-01-19 02:49:17
金秋(2017年4期)2017-06-07 08:22:16
中國材料進展(2016年10期)2016-12-26 06:50:20
濰坊學院學報(2016年2期)2016-12-01 13:00:11
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
新聞傳播(2015年11期)2015-07-18 11:15:04
