ADAM10抑制劑GI254023X對乳腺癌細胞體外生物學行為的影響及機制
2017-11-14 05:24:33徐杰劉佳錢龍娣杜秀平韓正祥
山東醫藥 2017年38期
關鍵詞:乳腺癌
徐杰,劉佳,錢龍娣,杜秀平,韓正祥
(1泰山醫學院附屬醫院,山東泰安 271000;2泰安市中心醫院;3徐州醫科大學附屬醫院)
ADAM10抑制劑GI254023X對乳腺癌細胞體外生物學行為的影響及機制
徐杰1,劉佳2,錢龍娣3,杜秀平3,韓正祥3
(1泰山醫學院附屬醫院,山東泰安 271000;2泰安市中心醫院;3徐州醫科大學附屬醫院)
目的觀察高選擇性去整合素金屬蛋白酶10(ADAM10)抑制劑GI254023X對乳腺癌細胞株MCF-7、MDA-MB-231增殖、凋亡、侵襲、遷移等生物學行為的影響,并初步探討其可能作用機制。方法取乳腺癌細胞株MCF-7、MDA-MB-231,用不同濃度(0、10、20、30、40、50 μmol/L)的GI254023X分別處理兩種細胞48、72 h,采用CCK-8檢測細胞增殖能力。取MCF-7、MDA-MB-231細胞,將其分為對照組、低濃度組與高濃度組,低濃度組加入10 μmol/L的GI254023X,高濃度組加入20 μmol/L的GI254023X,對照組加入和實驗組等量的DMSO溶劑,采用流式細胞儀測算細胞凋亡率,Transwell實驗檢測細胞遷移侵襲能力,Western boltting法檢測切割后的Notch1(Cleaved-Notch1)、Hes-1蛋白表達,qRT-PCR法檢測細胞Hes-1 mRNA表達。結果隨著GI254023X藥物濃度增加及作用時間的延長,MCF-7、MDA-MB-231細胞增殖抑制率不斷升高(P均<0.05)。與對照組相比,低濃度組、高濃度組MCF-7、MDA-MB-231細胞凋亡率高,遷移細胞數、侵襲細胞數少,Cleaved-Notch1、Hes-1蛋白表達少,Hes-1 mRNA表達少,且高濃度組較低濃度組更明顯(P均<0.05)。結論ADAM10抑制劑GI254023X可抑制乳腺癌細胞株MCF-7、MDA-MB-231增殖,誘導其凋亡,減弱其遷移和侵襲運動能力,該作用可能是通過下調ADAM10的表達來抑制Notch1信號通路的活化實現的。
去整合素金屬蛋白酶10;乳腺腫瘤;細胞增殖;細胞凋亡;細胞遷移;細胞侵襲;Notch1信號通路
乳腺癌是發生在乳腺上皮組織的惡性腫瘤,是全球女性發病率最高的惡性腫瘤之一,占女性新發惡性腫瘤的29%,其病死率僅次于肺癌而位居第二[1],給女性患者的生活及生命帶來了極為嚴重的影響。去整合素金屬蛋白酶(ADAMs)因分子結構上有解聚素域和金屬蛋白酶域兩個重要功能的結構域而命名,是一個新的基因家族。ADAMs家族成員在細胞黏附、細胞遷移、白細胞募集、炎癥、蛋白水解等方面發揮各自的作用[2]。ADAM10是ADAMs家族重要成員之一,干擾后可抑制膀胱癌、舌鱗癌等腫瘤細胞的增殖、遷移及侵襲能力[3~5]。Pruessmeyer等[6]發現,ADAM10在乳腺癌組織中過表達。抑制ADAM10表達是否也會抑制體外乳腺癌細胞的增殖、遷移與侵襲并促進其凋亡,尚未見報道。因此,2015年1~6月,我們觀察了ADAM10抑制劑GI254023X對人乳腺癌細胞生物學行為的影響,并探討其機制。
1 材料與方法
1.1 細胞及試劑 人乳腺癌細胞株MCF-7、MDA-MB-231購自中國科學院上海細胞庫。DMEM培養基、RPMI1640培養基及胎牛血清購自美國Gibco公司;GI254023X購自北京歐凱納斯科技有限公司;二甲基亞砜(DMSO)購自美國Sigma公司;CCK-8細胞增殖試驗試劑盒購自日本同仁化學研究所;Annexin V-APC/7-AAD試劑盒購自南京凱基生物科技發展有限公司;M-MLV反轉錄酶和總RNA提取試劑TRIzol購自美國Invitrogen公司;熒光定量PCR試劑盒購自美國Bio-Rad公司;Western blotting一抗購自Cell Signalling Technology公司。
1.2 細胞培養 將MCF-7常規培養在含有10%胎牛血清的DMEM培養液中,MDA-MB-231細胞常規培養在含有10%胎牛血清的RPMI1640培養液中,均置于37 ℃、5% CO2、飽和濕度的培養箱中培養。
1.3 細胞增殖抑制率測算 取對數生長期生長狀態良好的MCF-7、MDA-MB-231細胞,計數后調整細胞懸液至細胞密度為1.0×105/mL,按每孔100 μL接種于96孔板,培養24 h。24 h后實驗組分別加入10、20、30、40、50 μmol/L的GI254023X處理細胞,對照組加入和實驗組等量的DMSO溶劑,每孔設3個復孔。接種后細胞置于37 ℃、5% CO2飽和濕度培養箱中分別培養48、72 h后,每孔加入10 μL CCK-8試劑,繼續孵育4 h后用酶標儀檢測波長450 nm處的OD值。以藥物濃度為橫坐標,增殖抑制率為縱坐標,繪制細胞生長抑制曲線。細胞增殖抑制率=[1-(實驗組OD值/對照組OD值)]×100%。
1.4 細胞凋亡率測算 將處于對數生長期的MCF-7、MDA-MB-231細胞接種于6孔板上,分為對照組、低濃度組、高濃度組。低濃度組加入10 μmol/L的GI254023X,高濃度組加入20 μmol/L的GI254023X,對照組加入等量的DMSO溶劑。孵育48 h后,2 000 r/min離心5 min收集細胞。用PBS洗滌細胞2次,每(1~5)×105個細胞加入500 μL 1×binding buffer重懸細胞。加入5 μL AnnexinV-APC,混勻,室溫避光孵育15 min,加入5 μL 7-AAD混勻后冰浴避光上流式細胞儀分析,計算細胞凋亡率。
1.5 細胞遷移能力觀察 采用Transwell侵襲小室試驗。細胞處理及分組同1.4,常規洗滌、消化、離心,收集細胞制成細胞懸液,用無血清的培養基調整細胞密度為1.0×106/mL,接種于Transwell小室的上室150 μL,下室為10%胎牛血清的培養基600 μL。培養24 h后,將Transwell小室取出,用棉簽輕輕擦去上室表面黏附的細胞,95%甲醇固定20 min,PBS洗3遍,結晶紫染色30 min。使用倒置顯微鏡,觀測上室遷移至微孔膜下層的細胞,拍攝5個隨機視野,并進行細胞計數。
1.6 細胞侵襲能力觀察 Transwell侵襲小室外膜涂50 μL纖維黏連蛋白,用槍頭攤平,風干后內膜平鋪Matrigel稀釋液30 μL,37 ℃過夜凝膠。細胞處理及分組同1.4,常規洗滌、消化、離心,收集細胞制成細胞懸液,用無血清的培養基調整細胞密度為1×106/mL,其他步驟同遷移實驗。
1.7 切割后的Notch1(Cleaved-Notch1)、Hes-1蛋白檢測 細胞處理及分組同1.4。收集處理48 h的細胞,提取各組蛋白,BCA法測蛋白濃度,經SDS-PAGE電泳分離后,將蛋白轉移至NC膜上,明膠室溫封閉1 h。加入一抗抗體(Cleaved-Notch1 1∶1 000, Hes-1 1∶1 000, β-actin 1∶1 000),4 ℃孵育過夜,1×PBST液洗膜3次,10 min/次。后加入HRP標記的山羊抗兔IgG,室溫孵育1 h,PBST洗膜3次后,加入顯色液(BIO-RAD),采用生物分子成像儀LAS4000(GE Healthcare公司)成像。內參照選用β-actin,結果以Image J分析處理,計算得出相對灰度值來代表Cleaved-Notch1、Hes-1蛋白相對表達量。
1.8 Hes-1 mRNA檢測 采用RT-qPCR法。細胞處理及分組同1.4。收集細胞,按TRIzol總RNA分離試劑盒說明書(Invitrogen公司)進行總RNA提取,cDNA的合成按照M-MLV逆轉錄酶說明書(Invitrogen 公司)推薦的條件。采用LightCycler 480 SYBR Green Ⅰ Master熒光定量PCR試劑盒說明加入相應組分,反應體系為20 μL。Hes-1上、下游引物分別為5′-AACACTGATTTTGGATGCTCTGA-3′、5′-GGTACTTCCCCAGCACACTTG-3′,GAPDH上、下游引物分別為5′-CATCAAGAAGGTGGTGAAGCAG-3′、5′-CAAAGGTGGAGGAGTGGGTG-3′。實時定量PCR循環參數:95 ℃變性10 s,60 ℃退火,延伸60 s,共40個循環。以GAPDH為內參,采用2-ΔΔCt法計算Hes-1 mRNA的相對表達量。獨立實驗重復3次。
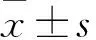
2 結果
2.1 不同濃度GI254023X處理MCF-7、MDA-MB-231細胞增殖抑制率比較 見表1、2。

表1 不同濃度GI254023X處理MCF-7細胞增殖抑制率比較
注:與同濃度48 h相比,aP<0.05;與10 μmol/L相比,bP<0.05;與20 μmol/L相比,cP<0.05;與30 μmol/L相比,dP<0.05;與40 μmol/L相比,eP<0.05。
表2 不同濃度GI254023X處理MDA-MB-231細胞增殖抑制率比較
注:與同濃度48 h相比,aP<0.05;與10 μmol/L相比,bP<0.05;與20 μmol/L相比,cP<0.05;與30 μmol/L相比,dP<0.05;與40 μmol/L相比,eP<0.05。
2.2 各組細胞凋亡率比較 見表3。
表3 各組細胞凋亡率比較
注:與對照組相比,*P<0.05;與低濃度組相比,#P<0.05。
2.3 各組細胞遷移能力比較 見表4。

表4 各組細胞遷移能力比較(個
注:與對照組相比,*P<0.05;與低濃度組相比,#P<0.05。
2.4 各組細胞侵襲能力比較 見表5。
表5 各組細胞侵襲能力比較(個
注:與對照組相比,*P<0.05;與低濃度組相比,#P<0.05。
2.5 各組細胞Cleaved-Notch1、Hes-1表達比較 見表6。
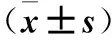
表6 各組細胞Cleaved-Notch1、Hes-1表達比較
注:與對照組相比,*P<0.05;與低濃度組相比,#P<0.05。
3 討論
ADAMs家族是一個新的基因家族,屬Ⅰ型跨膜蛋白,其在調節細胞-細胞和細胞-基質的相互作用中至關重要。ADAM10為該家族重要成員之一,其可引起多種細胞表面分子脫落,包括鈣黏素和淀粉樣前體蛋白。ADAM10能夠調節腫瘤微環境和腫瘤進展的重要過程,包括細胞增殖、遷移和血管生成等。研究[7,8]已經證實,過表達的ADAM10是腫瘤高復發、高轉移風險的一項潛在預測指標。本研究結果發現,10、20、30、40、50 μmol/L的ADAM10抑制劑GI254023X處理乳腺癌細胞株MCF-7、MDA-MB-231細胞增殖抑制率不斷升高,兩兩相比差異有統計學意義,且同濃度時72 h較48 h細胞增殖抑制率高,表明GI254023X可抑制MCF-7、MDA-MB-231的增殖,具有明顯的誘導凋亡作用,并且隨濃度的增高或時間的延長而增強。侵襲性是腫瘤細胞的重要惡性表型,研究腫瘤細胞的侵襲能力對于臨床腫瘤疾病轉移和復發的治療具有重要價值。本實驗結果還顯示,與對照組比較,低濃度組、高濃度組MCF-7、MDA-MB-231細胞凋亡率高,遷移細胞數、侵襲細胞數少,且高濃度組較低濃度組更明顯。可見,GI254023X對于MCF-7、MDA-MB-231細胞的生長起到抑制作用,且能拮抗腫瘤細胞的遷移及侵襲轉移。
Notch信號通路是決定細胞命運的重要通路[9],而ADAM10在Notch1蛋白的切割中起重要作用,參與Notch1靶基因Hes-1及v-Myc的轉錄激活[10~12]。有研究[13]發現,下調Notch1和Jagged1的表達可抑制子宮頸癌和絨毛膜癌細胞的侵襲。在非小細胞肺癌細胞及急性T淋巴細胞白血病細胞中沉默ADAM10的表達,可觀察到細胞的增殖、遷移及侵襲能力均受到抑制,Notch1的活化受到明顯抑制,提示該作用可能與抑制Notch1信號通路的活化有關[14,15]。本研究發現,與對照組比較,低濃度組、高濃度組MCF-7、MDA-MB-231細胞Cleaved-Notch1、Hes-1蛋白表達少,且高濃度組較低濃度組更明顯。說明G1254023X抑制ADAM10后,MCF-7、MDA-MB-231細胞中Notchl的切割活化受到抑制。本研究發現,與對照組比較,低濃度組、高濃度組MCF-7、MDA-MB-231細胞Hes-1 mRNA表達少,且高濃度組較低濃度組更明顯,進一步證實G1254023X可抑制MCF-7、MDA-MB-231細胞Notchl信號通路的活化。
綜上所述,ADAM10抑制劑GI254023X具有明顯影響乳腺癌細胞株MCF-7、MDA-MB-231生物學行為的作用,具體表現為增殖抑制、誘導凋亡,并且減弱遷移和侵襲運動能力。GI254023X可能通過下調ADAM10的表達,抑制Notch1信號通路的活化,從而對乳腺癌細胞的生物學行為產生影響。
[1] Siegel R, Ma J, Zou Z, et al. Cancer statistics, 2014[J]. CA Cancer J Clin, 2014,64(1):9-29.
[2] Dreymueller D, Pruessmeyer J, Groth E, et al. The role of ADAM-mediated shedding in vascular biology[J]. Eur J Cell Biol, 2012,91(6-7):472-485.
[3] Armanious H, Gelebart P, Anand M, et al. Constitutive activation of metalloproteinase ADAM10 in mantle cell lymphoma promotes cell growth and activates the TNFalpha/NFkappaB pathway[J]. Blood, 2011,117(23):6237-6246.
[4] Fu L, Liu N, Han Y, et al. ADAM10 regulates proliferation, invasion, and chemoresistance of bladder cancer cells[J]. Tumour Biol, 2014,35(9):9263-9268.
[5] Shao Y, Sha XY, Bai YX, et al. Effect of A disintegrin and metalloproteinase 10 gene silencing on the proliferation, invasion and migration of the human tongue squamous cell carcinoma cell line TCA8113[J]. Mol Med Rep, 2015,11(1):212-218.
[6] Pruessmeyer J, Ludwig A. The good, the bad and the ugly substrates for ADAM10 and ADAM17 in brain pathology, inflammation and cancer[J]. Semin Cell Dev Biol, 2009,20(2):164-174.
[7] Lee SB, Schramme A, Doberstein K, et al. ADAM10 is upregulated in melanoma metastasis compared with primary melanoma[J]. J Invest Dermatol, 2010,130(3):763-773.
[8] Xu Q, Liu X, Chen W, et al. Inhibiting adenoid cystic carcinoma cells growth and metastasis by blocking the expression of ADAM 10 using RNA interference[J]. J Transl Med, 2010,8:136.
[9] Sharma A, Paranjape AN, Rangarajan A, et al. A monoclonal antibody against human Notch1 ligand-binding domain depletes subpopulation of putative breast cancer stem-like cells[J]. Mol Cancer Ther, 2012,11(1):77-86.
[10] Ma J, Tang X, Wong P, et al. Noncanonical activation of Notch1 protein by membrane type 1 matrix metalloproteinase (MT1-MMP) controls melanoma cell proliferation[J]. J Biol Chem, 2014,289(12):8442-8449.
[11] Tohda S. NOTCH signaling roles in acute myeloid leukemia cell growth and interaction with other stemness-related signals[J]. Anticancer Res, 2014,34(11):6259-6264.
[12]. Capaccione KM, Pine SR. The Notch signaling pathway as a mediator of tumor survival[J]. Carcinogenesis, 2013,34(7):1420-1430.
[13] Pang RT, Leung CO, Ye TM, et al. MicroRNA-34a suppresses invasion through downregulation of Notch1 and Jagged1 in cervical carcinoma and choriocarcinoma cells[J]. Carcinogenesis, 2010,31(6):1037-1044.
[14] Guo J, He L, Yuan P, et al. ADAM10 overexpression in human non-small cell lung cancer correlates with cell migration and invasion through the activation of the Notch1 signaling pathway[J]. Oncol Rep, 2012,28(5):1709-1718.
[15] Sulis ML, Saftig P, Ferrando AA. Redundancy and specificity of the metalloprotease system mediating oncogenic NOTCH1 activation in T-ALL[J]. Leukemia, 2011,25(10):1564-1569.
10.3969/j.issn.1002-266X.2017.38.012
R737.9
A
1002-266X(2017)38-0040-04
江蘇省“六大人才高峰”B類項目(2014-WSW-040)。
韓正祥(E-mail: cnhzxyq@163.com)
2017-06-24)
猜你喜歡
中老年保健(2022年6期)2022-08-19 01:41:48
現代臨床醫學(2022年1期)2022-02-12 02:04:58
甘肅科技(2020年20期)2020-04-13 00:30:42
中國生殖健康(2019年2期)2019-08-23 08:11:42
中國生殖健康(2019年6期)2019-01-06 09:20:12
中國生殖健康(2019年5期)2019-01-06 09:16:40
幸福家庭(2019年14期)2019-01-06 09:15:38
祝您健康(2018年5期)2018-05-16 17:10:16
癌癥進展(2016年9期)2016-08-22 11:33:20
中國組織化學與細胞化學雜志(2016年4期)2016-02-27 11:16:08
