人肝細胞癌細胞DNA甲基化譜的檢測及分析
2017-12-19 09:06:44孫寧張佳林張城碩周翔宇陳保民
中國醫科大學學報 2017年12期
孫寧,張佳林,張城碩,周翔宇,陳保民
(中國醫科大學附屬第一醫院肝膽外科暨器官移植科,沈陽 110001)
人肝細胞癌細胞DNA甲基化譜的檢測及分析
孫寧,張佳林,張城碩,周翔宇,陳保民
(中國醫科大學附屬第一醫院肝膽外科暨器官移植科,沈陽 110001)
目的檢測人肝細胞癌細胞的DNA甲基化譜,明確肝細胞癌細胞中差異甲基化位點和基因的表達分布情況,進一步探討DNA異常甲基化與肝細胞癌發生發展的關系。方法使用DNA甲基化芯片(Infinium Human Methylation 450K BeadChip)檢測人肝細胞癌細胞Huh7和人永生化肝細胞L02的甲基化譜,并對檢測結果進行生物學分析。結果共檢測到差異性甲基化位點102 254個,差異性甲基化基因26 511個,甲基化相關信號通路43個,其中57.3%的高甲基化CpG位點和39.4%的低甲基化CpG位點的甲基化差異程度≥50%,篩選后確定了3 222個顯著高甲基化基因和2 204個顯著低甲基化基因。結論在Huh7和L02細胞中存在大量差異性甲基化CpG位點及基因,Huh7細胞中可檢測到大量抑癌基因DNA的異常高甲基化,提示DNA異常甲基化與肝細胞癌的發生發展密切相關。
肝細胞癌; 甲基化譜; 生物信息學分析
原發性肝癌(primary liver cancer,PLC)是我國常見的消化系統惡性腫瘤,2015年我國PLC新發病例約為46.6萬人,發病率位列全部惡性腫瘤第4位,全年死亡病例約為42.2萬,致死率位列全部惡性腫瘤第3位[1]。肝細胞癌(hepatocellular carcinoma,HCC)是PLC最常見的類型,導致HCC發生發展的因素有很多,如慢性病毒性肝炎、酒精性肝硬化、黃曲霉素的攝入等,但其具體發病機制仍不明確,因此,研究其發生發展機制對尋找其新的治療方案至關重要。
隨著基因組領域研究的不斷深入,表觀遺傳學改變對疾病的影響逐漸引起研究者的關注。DNA異常甲基化是表觀遺傳學中最常見也是最重要的修飾,其異常改變被認為是導致包括HCC在內的多種腫瘤發生發展的主要因素[2]。研究[3-4]發現,基因的異常甲基化可影響其轉錄活性,使其表達下調,尤其是抑癌基因的異常高甲基化常可引起其抑癌功能的失活,最終導致腫瘤的發生。
高通量甲基化芯片Infinium Human Methylation 450K BeadChip能夠對人全基因組45萬個甲基化位點進行檢測,覆蓋了上一代27K甲基化芯片90%的位點,并同時增加了CpG島以外的CpG位點、人類干細胞非CpG甲基化位點、正常組織與腫瘤(多種癌癥)組織差異甲基化位點、編碼區以外的CpG島、miRNA啟動子區域和已通過GWAS的疾病相關區域的位點。因此,本研究使用Infinium Human Methylation 450K BeadChip對人HCC細胞系Huh7和人永生化肝細胞系L02的DNA甲基化譜進行檢測并對其進行分析,篩選出具有顯著差異的甲基化CpG位點和基因,明確HCC細胞中差異甲基化CpG位點的表達和分布情況,進一步探討DNA異常甲基化與HCC發生發展的關系。
1 材料與方法
1.1 材料
人永生化肝細胞L02、人HCC細胞系Huh7均購自中國科學院細胞庫;DNA提取試劑盒(QIAamp DNA Micro Kit)購自美國QIAGEN公司;甲基化修飾試劑盒購自沈陽百創特公司;人全基因組甲基化芯片(Infinium Human Methylation 450K BeadChip) 購自美國Illumina公司。
1.2 細胞培養
L02細胞及Huh7細胞分別單層貼壁生長于RPMI-1640培養液或DMEM培養液中,培養液中均含10%胎牛血清、青霉素G (100 U/L)、鏈霉素(100 μ g/L),于37 ℃、5% CO2恒溫密閉式培養箱中培養,隔日換液,取對數生長期細胞進行后續實驗。
1.3 DNA提取及甲基化修飾
用0.25%含EDTA胰酶消化細胞,收集細胞沉淀,取20 μ L Proteinase K 加入到1.5 mL Eppendorf管內,按DNA提取試劑盒說明書中的操作步驟提取Huh7、L02細胞DNA,Nano Drop核酸蛋白分析儀檢測DNA提取純度和濃度,若OD260nm/OD280mn在1.7~2.0之間則符合實驗要求,否則樣品需要重新提取。將提取的Huh7、L02細胞DNA按照甲基化修飾試劑盒說明書中的操作步驟進行甲基化修飾。
1.4 芯片雜交、掃描及數據分析
按照Illumina公司提供的步驟進行芯片雜交;數據掃描使用manifest文件,采用Illumina公司官方提供的Methylation Module of GenomeStudio software using Methylation v1.9軟件對芯片數據進行分析。β-value [β = intensity of the methylated allele(M)/(intensity of the unmethylated allele + intensity of the methylated allele + 100)]代表CpG位點的甲基化水平,數值越大,越趨近于1,甲基化程度越高[5];β-difference (絕對值)為表達差異性分數。本研究使用empirical bayes moderated t 檢驗及Bonferroni校正來識別Huh7細胞和L02細胞之間的差異性甲基化CpG位點。初步篩選差異性甲基化CpG位點的納入標準為經過校正的P值(Adjust P)≤0.05,同時表達差異性分數(|β-difference|)≥0.2,被定義為具有統計學意義。而Adjust P > 0.05和CpG覆蓋<95%則被排除。另外,為進一步篩選具有顯著差異的甲基化CpG位點,本研究使用了額外的過濾標準: (1) Adjust P ≤0.05,P ≤1.06×10-7;(2)顯著的高甲基化位點,即差異性甲基化水平>20%,L02甲基化水平<25%;(3)顯著低甲基化位點,即差異性甲基化水平>20%,Huh7甲基化水平<25%。
2 結果
2.1 Huh7細胞全基因組差異性甲基化位點表達情況
本研究中,共檢測到差異性甲基化位點102 254個,差異性甲基化基因26 511個。其中,高甲基化CpG位點62 702個(61.3%),包含12 665個高甲基化基因;低甲基化位點39 552個(38.7%),包含13 846個低甲基化基因。另外,在CpG島中,檢測到了差異性CpG位點41 178個(40.3%);在CpG shores檢測到了差異性甲基化CpG位點21 150個(20.7%);在CpG shelves檢測到9 030個(8.8%)差異性甲基化CpG位點。見表1,圖1。
2.2 差異性甲基化CpG位點甲基化差異程度的分布
本研究結果顯示,有35 937個(57.3%)高甲基化CpG位點和15 587個(39.4%)低甲基化CpG位點的甲基化差異程度≥50%;18 529個(29.5%)高甲基化CpG位點和14 177個(35.8%)低甲基化CpG位點的甲基化差異程度≥30%但<50%;8 236個(13.1%)高甲基化CpG位點和9 788個(24.8%)低甲基化CpG位點的甲基化差異程度<30%但≥20%。見表2。

表1 總體差異性甲基化CpG位點分布 [n(%)]Tab.1 Distribution of all the differentially methylated CpG sites [n(%)]
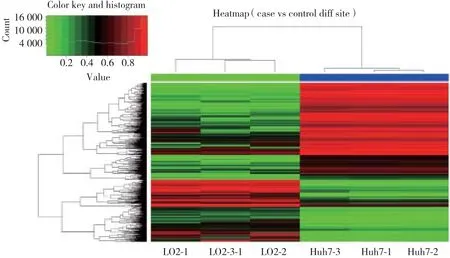
圖1 差異性甲基化CpG位點的聚類分析Fig.1 Hierarchical cluster analysis of all differentially methylated CpG sites between Huh7 cells and L02 cells
2.3 顯著差異性甲基化CpG位點分布及差異性甲基化基因篩選
通過使用嚴格的篩選條件對差異性甲基化CpG位點進行篩選后,確定了5 285個明顯的高甲基化CpG位點(包括3 222個基因)和2 659個明顯的低甲基化CpG位點(包括2 204個基因)。見表3~5,圖2。
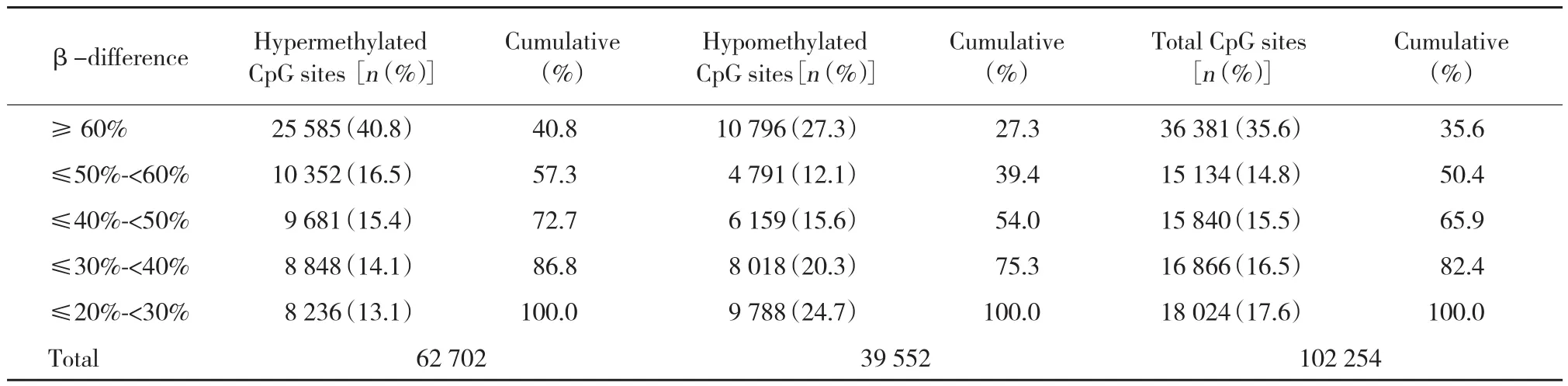
表2 差異性甲基化CpG位點甲基化差異程度的分布Tab.2 Distribution of all the differentially methylated CpG sites between Huh7 and L02 cells based on methylation status
2.4 差異性甲基化基因的GO富集分析和KEGG通路分析
本研究使用GO富集分析(http://www.geneontol ogy.org/)和 KEEG 通路分析 (the Kyoto Encyclopedia of Genes and Genomes,http://www.genome.jp/kegg/)對差異性位點進行相關通路分析。GO富集的結果如下:2 107個差異甲基化基因富集于9個生化過程相關功能集,富集最明顯的包括負調控的細胞增殖,子宮內的胚胎發育等;13 351個差異甲基化基因富集于17個分子功能相關功能集,富集最明顯的包括蛋白結合,金屬離子結合等;18 041個差異甲基化基因富集于與細胞成分有關的功能集,富集最明顯的主要是細胞核、細胞質等。KEGG通路分析顯示43個信號通路涉及5 195個差異甲基化的基因,這些基因主要富集于代謝通路、癌癥相關通路、MAPK信號通路、Wnt 信號通路、VEGF 信號通路和 p53 信號通路等,見表6。
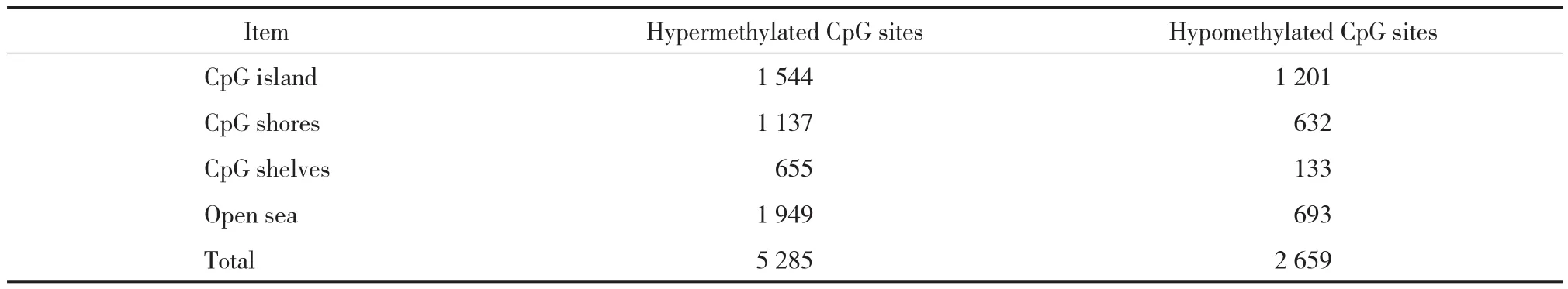
表3 顯著差異性甲基化位點分布情況(n)Tab.3 Distribution of significantly differentially methylated CpG sites in Huh7 cells when compared with L02 cells(n)
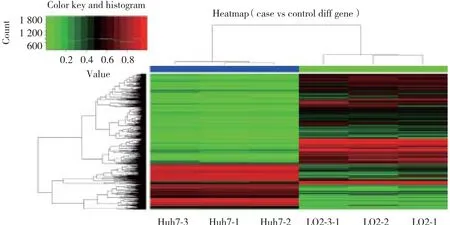
圖2 差異性甲基化CpG基因的聚類分析Fig.2 Hierarchical cluster analysis of all differentially methylated genes between Huh7 cells and L02 cells
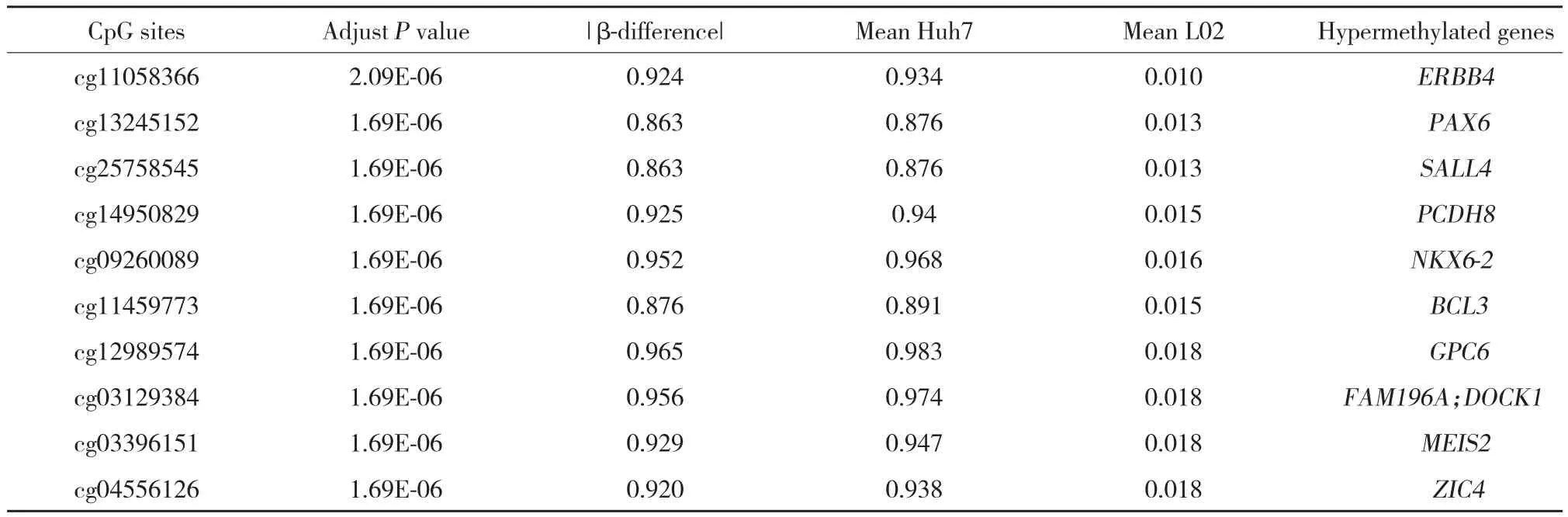
表4 前10個顯著差異高甲基化基因Tab.4 Top 10 significant hypermenthylated CpG sites and genes within DMRs in Huh7 cells compared with L02 cells
3 討論
近年來,基因的DNA異常甲基化被認為是導致多種腫瘤發生的重要因素[6-7]。既往針對HCC相關基因的DNA甲基化研究雖然能夠為HCC的診斷和預后評估提供一些新的甲基化標記物,但關于HCC DNA甲基化譜的研究甚少。因此,本研究利用Infinum Human Methylation 450K芯片技術對HCC細胞的DNA甲基化譜進行檢測,并對結果進行分析。
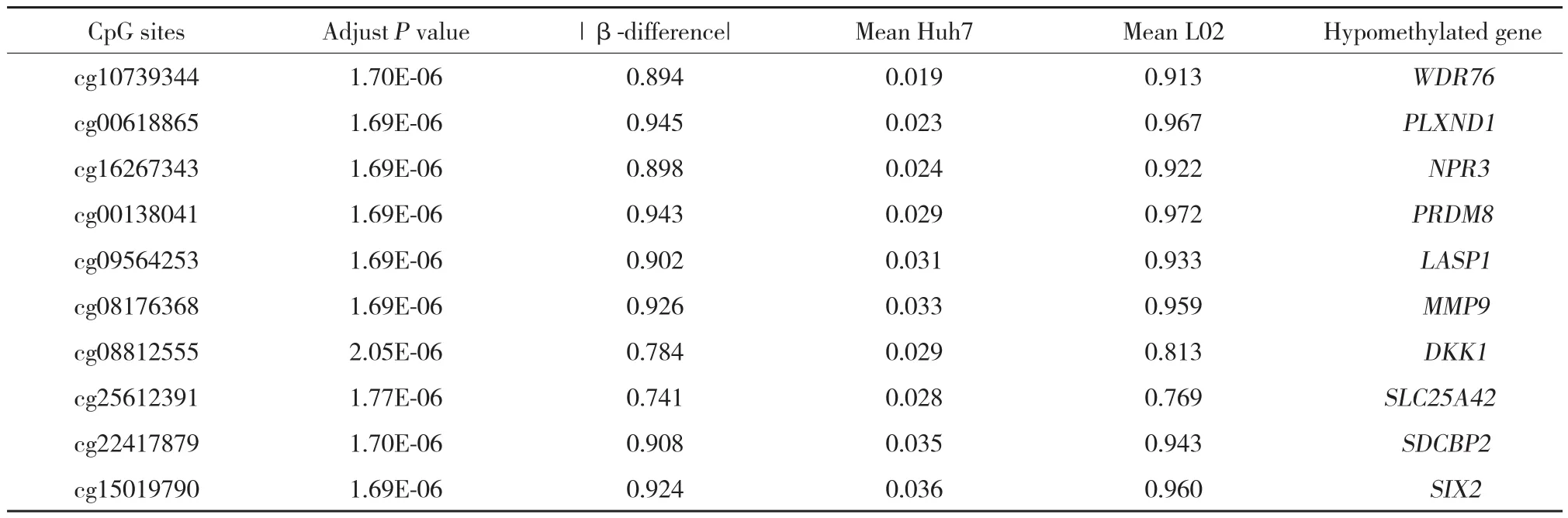
表5 前10個顯著差異低甲基化基因Tab.5 Top 10 significant CpG hypomethylated sites and genes within DMRs in Huh7 cells compared with L02 cells
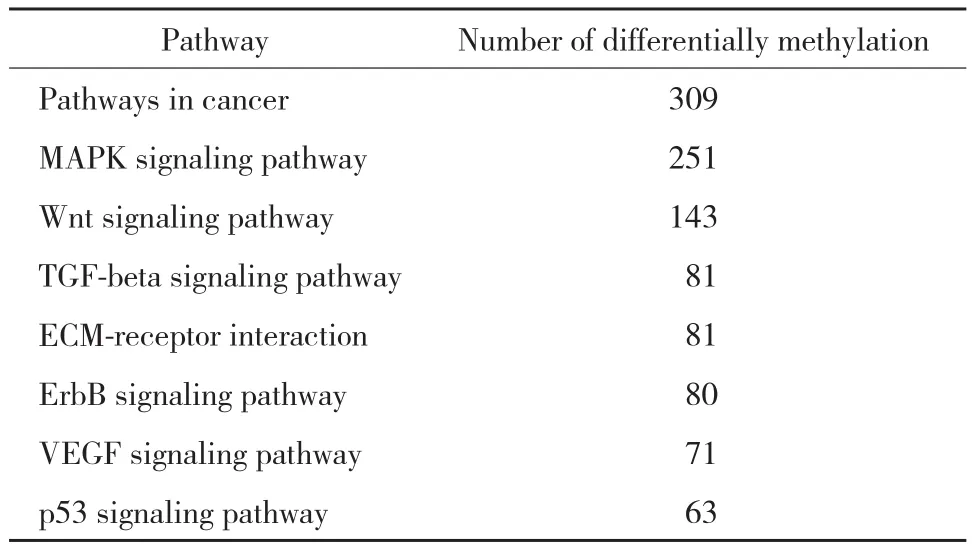
表6 主要HCC相關通路中差異甲基化基因數目Tab.6 Number of differentially methylated gene in HCC related pathway
Infinium Human Methylation 450K甲基化芯片涵蓋99%在注釋的RefSeq基因啟動子區的多個位點、5’-UTR、第一外顯子、3’-UTR等重要區域[8]。在本研究結果中,共檢測到102 254個差異性甲基化CpG位點,并且高甲基化CpG位點(62 702,61.3%)明顯多于低甲基化CpG位點(39 552,38.7%),表明在Huh7細胞中DNA異常甲基化是一種常見事件,而且發生頻率較高。同時,還發現顯著高甲基化CpG位點在CpG島的分布要多于顯著低甲基化位點(1 544 vs.1 201),這與之前其他相關的DNA甲基化基因分析研究[9-12]結果一致。
有研究[13-17]認為,DNA異常甲基化不僅發生在CpG島,也可能發生在CpG shores和CpG shelves等區域,并同樣能夠導致抑癌基因的功能失活。本研究還發現在CpG shores的確分布有一定數量的顯著差異CpG位點,且高甲基化CpG位點多于低甲基化位點(1 137 vs 632),而在CpG shelves,高甲基化CpG位點更明顯多于低甲基化CpG位點(655 vs 133)。本研究結果不僅進一步證實了其他研究的結論,同時也提示,在HCC細胞中能夠檢測到大量DNA異常高甲基化位點,這些異常位點很可能與HCC相關抑癌基因功能失活有關,參與了HCC的發生發展。
另外,本研究通過對差異性甲基化基因的GO分析和KEGG通路分析發現DNA的異常甲基化能夠影響細胞周期相關蛋白p16(Ink4a)、p53、TGF-β/SMAD信號等經典癌癥相關通路[18]。在本研究中,雖然對HCC細胞DNA的甲基化譜進行了檢測及分析,但是仍需要進一步的實驗研究對現有的芯片結果進行驗證,繼續深入探討DNA異常甲基化在HCC發生發展中所起的作用及其臨床意義。現有研究[19-20]已證實,在HCC中,Wnt通路的激活與抑癌基因DNA異常高甲基化有關,Erb受體信號途徑和MAPK信號通路也已被證實其相關蛋白質經過表觀遺傳學異常修飾后活性改變,從而導致了腫瘤生長和轉移[16,21],這些研究對下一步芯片結果的驗證均具有一定的指導意義。
綜上所述,本研究應用高通量甲基化芯片測序技術對Huh7細胞和L02細胞的全基因組DNA甲基化模式進行了探討,初步明確了差異性甲基化CpG位點/區域/基因在HCC細胞中的表達頻率和分布情況,為今后HCC相關的甲基化研究提供了更多信息。本研究結果中的顯著差異位點和基因仍需進行驗證,其相關分子機制也有待進一步研究,以繼續深入探究DNA甲基化與HCC發生發展的關系,也為今后HCC的去甲基化治療奠定基礎。
[1] CHEN W,ZHENG R,BAADE PD,et al. Cancer statistics in China,2015[ J]. CA Cancer J Clin,2016,66(2):115-132. DOI:10.3322/caac.21338.
[2] KULIS M,ESTELLER M. DNA methylation and cancer[ J]. Adv Genet,2010,70:27-56. DOI:10.1016/B978-0-12-380866-0.60002-2.
[3] HERMAN JG,BAYLIN SB. Gene silencing in cancer in association with promoter hypermethylation[ J]. N Engl J Med,2003,349( 21):2042-2054. DOI:10.1056/NEJMra023075.
[4] ESTELLER M. Epigenetic gene silencing in cancer:the DNA hypermethylome[ J]. Hum Mol Genet,2007,16( 1):R50-R59. DOI:10.1093/hmg/ddm018.
[5] BIBIKOVA M,LE J,BARNES B,et al. Genome-wide DNA methylation profiling using Infinium(R) assay[ J]. Epigenomics,2009,1( 1):177-200. DOI:10.2217/epi.09.14.
[6] JONES P A,TAKAI D. The role of DNA methylation in mammalian epigenetics[ J]. Science,2001,293( 5532):1068-1070. DOI:10.1126/science.1063852.
[7] ESTELLER M. Epigenetics in cancer[ J]. N Engl J Med,2008,358(11):1148-1159. DOI:10.1056/NEJMra072067.
[8] BIBIKOVA M,BARNES B,TSAN C,et al. High density DNA methylation array with single CpG site resolution[ J]. Genomics,2011,98(4):288-295. DOI:10.1016/j.ygeno.2011.07.007.
[9] GAO W,KONDO Y,SHEN L,et al. Variable DNA methylation patterns associated with progression of disease in hepatocellular carcinomas[ J]. Carcinogenesis,2008,29( 10):1901-1910. DOI:10.1093/carcin/bgn170.
[10] SHIN SH,KIM BH,JANG JJ,et al. Identification of novel methylation markers in hepatocellular carcinoma using a methylation array[J]. J Korean Med Sci,2010,25( 8):1152-1159. DOI:10.3346/jkms.2010.25.8.1152.
[11] AMMERPOHL O,PRASTCHKE J,SCHAFMAYER C,et al. Distinct DNA methylation patterns in cirrhotic liver and hepatocellular carcinoma[ J]. Int J Cancer,2012,130( 6):1319-1328. DOI:10.1002/ijc.26136.
[12] KOHLES N,NAGEL D,JUNGST D,et al. Prognostic relevance of oncological serum biomarkers in liver cancer patients undergoing transarterial chemoembolization therapy[ J]. Tumour Biol,2012,33(1):33-40. DOI:10.1007/s13277-011-0237-7.
[13] YATES DR,REHMAN I,MEUTH M,et al. Methylational urinalysis:a prospective study of bladder cancer patients and age stratified benign controls[ J]. Oncogene,2006,25( 13):1984-1988. DOI:10.1038/sj.onc.1209209.
[14] DUZIEC E,MIAH S,CHOUDHRY HM,et al. Hypermethylation of CpG islands and shores around specific microRNAs and mirtrons is associated with the phenotype and presence of bladder cancer,clinical cancer research[ J]. Clin Cancer Res,2011,17( 6):1287-1296.DOI:10.1158/1078-0432.CCR-10-2017.
[15] DOI A,PARK IH,WEN B,et al. Differential methylation of tissueand cancer-specific CpG island shores distinguishes human induced pluripotent stem cells,embryonic stem cells and fibroblasts[ J]. Nat Genet,2009,41( 12):1350-1353. DOI:10.1038/ng.471.
[16] IRIZARRY RA,LADD-ACOSTA C,WEN B,et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores[ J]. Nat Genet,2009,41(2):178-186. DOI:10.1038/ng.298.
[17] OGOSHI K,HASHIMOTO S,NAKATANI Y,et al. Genome-wide profiling of DNA methylation in human cancer cells[ J]. Genomics,2011,98( 4):280-287. DOI:10.1016/j.ygeno.2011.07.003.
[18] KUO KK,JIAN SF,LI YJ,et al. Epigenetic inactivation of transforming growth factor-beta1 target gene HEYL,a novel tumor suppressor,is involved in the P53-induced apoptotic pathway in hepatocellular carcinoma[ J]. Hepatol Res,2015,45( 7):782-793. DOI:10.1111/hepr.12414.
[19] UMER M,QURESHI SA,HASHMI ZY,et al. Promoter hypermethylation of Wnt pathway inhibitors in hepatitis C virus-induced multistep hepatocarcinogenesis[ J]. Virol J,2014,11:117. DOI:10.1186/1743-422X-11-117.
[20] DING SL,YANG ZW,WANG J,et al. Integrative analysis of aberrant Wnt signaling in hepatitis B virus-related hepatocellular carcinoma[ J]. World J Gastroenterol,2015,21( 20):6317-6328. DOI:10.3748/wjg.v21.i20.6317.
[21] STEFANSKA B,CHEISHVILI D,SUDERMAN M,et al. Genome-wide study of hypomethylated and induced genes in patients with liver cancer unravels novel anticancer targets[ J]. Clin Cancer Res,2014,20( 12):3118-3132. DOI:10.1158/1078-0432.CCR-13-0283.
Exploring Genome-wide Profiles of DNA Methylation in Human Hepatocellular Carcinoma Cells via Bioinformatics Analysis
SUN Ning,ZHANG Jialin,ZHANG Chengshuo,ZHOU Xiangyu,CHEN Baomin
(Department of Hepatobiliary and Transplantation Surgery,The First Hospital,China Medical University,Shenyang 110001,China)
ObjectiveTo detect the genome-wide profiles of DNA methylation in human hepatocellular carcinoma (HCC) cells and to identify the distribution of differentially methylated sites and genes in order to explore the relationship between aberrant DNA methylation and hepatocellular carcinoma.MethodsThe Infinium Human Methylation 450K BeadChip was used to identify the genome-wide aberrant DNA methylation profiles in Huh7 and L02 cell lines.ResultsTotally 102 254 differentially methylated CpG sites and 26 511 genes,involving 43 signaling pathways,were detected when Huh7 and L02 cell lines were compared. The absolute β-difference in 57.3%of the hypermethylated CpG sites and 39.4% of the hypomethylated CpG sites was reported to be ≥ 50%. A total of 3 222 hypermethylated genes and 2 204 hypomethylated genes were identified.ConclusionWe detected many aberrant methylated sites and genes in HCC cells. The abnormal DNA methylation exhibits an important role in the occurrence and development of HCC.
hepatocellular carcinoma; methylation profiles; bioinformatics analysis
R735.7
A
0258-4646(2017)12-1111-06
http://kns.cnki.net/kcms/detail/21.1227.R.20171130.1810.022.html
10.12007/j.issn.0258-4646.2017.12.012
沈陽市科學技術計劃(F13-F13-212-9-00)
孫寧(1986-),女,醫師,博士.
張佳林,E-mail:jlzcmu@126.com
2017-03-28
網絡出版時間:2017-11-30 18:10
(編輯 王又冬)
猜你喜歡
音樂探索(2022年2期)2022-05-30 21:01:37
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
