兩階段設計在醫療器械非隨機臨床試驗中的應用
2018-01-03 01:30:20趙延延許毓君
中國衛生統計 2017年6期
關鍵詞:設計
趙延延 許毓君 王 楊 李 衛△
兩階段設計在醫療器械非隨機臨床試驗中的應用
趙延延1?許毓君2?王 楊1李 衛1△
目的介紹兩階段設計的概念、使用流程和注意事項,并以某外周血管支架臨床試驗為例介紹其在醫療器械非隨機臨床試驗中的應用。方法基于傾向性評分的兩階段設計能使醫療器械非隨機臨床試驗滿足隨機化和前瞻性的原則。我們以某外周血管支架的非劣效試驗為例,介紹兩階段設計的流程和細節。結果兩階段設計解決了非隨機臨床試驗由于非隨機所帶來的可能的研究偏倚及傾向性評分方法使用過程中的主觀不確定性,實現了對隨機臨床試驗的模擬,從而保障了非隨機臨床試驗研究設計和統計分析的客觀性和前瞻性。結論兩階段設計能夠增加臨床試驗的可行性,整合高質量的外部數據,縮短試驗周期,提高試驗效率,最終得到可靠的結論,具有較大的推廣價值。
兩階段設計 傾向性評分 醫療器械 非隨機臨床試驗
設計科學、實施嚴謹的隨機對照試驗是評價醫療產品安全性和有效性的金標準。然而出于倫理學或臨床可行性的考慮,許多醫療器械臨床試驗無法采用嚴格的隨機對照研究設計[1];此外,隨機對照試驗往往耗時很長,如醫療器械臨床試驗的周期是3~7年,而隨著醫療技術的發展,器械產品的更替周期要短于隨機對照試驗的周期,如心臟支架產品的生命周期約為2年。為了能夠使好產品盡早上市,造福于廣大病患,急需一種既能夠作為隨機臨床試驗補充,又符合統計學規范的研究設計方法。
非隨機臨床試驗(non-randomized clinical trial)是指在臨床試驗中受試者所分配的干預不是由機會決定,而由受試者或研究者指定[2],根據對照組的不同可以分為非隨機同期對照試驗和非隨機歷史對照試驗。相比于傳統的隨機對照試驗,這類試驗能夠克服實際研究開展過程中面臨的倫理學或可行性問題,整合高質量的外部數據,高效地回答臨床實踐問題,在國外已有不少被用于醫療器械上市前的申報中[3-4]。但非隨機臨床試驗本身缺少隨機化的環節,會導致組間基線變量不均衡,帶來效應估計的偏倚;同時結局評價不具有前瞻性,即研究者在獲得試驗結局數據之后再進行評分建模,通過大量重復的事后分析得到有利于試驗產品的結果,這使得試驗結果的可靠程度備受質疑,目前在國內尚無以非隨機對照臨床試驗的結果作為上市前確證性證據而獲得審批的產品。傾向性評分的方法本身雖然在解決隨機化問題時顯示出極大的優越性[5],但在臨床試驗中多被用于事后分析,存在數據導向、人為操控數據獲得陽性結果的嫌疑,分析結果通常亦不被認可。
兩階段設計是指在利用傾向性評分方法的基礎上,對試驗的流程進行人為的劃分[6],從而模擬試驗的隨機化過程,保障試驗設計和統計分析的前瞻性,進而獲得較為客觀可靠的試驗結果,被美國食品藥品監督管理局(FDA)推薦用于非隨機臨床試驗中[7]。本文將詳細介紹兩階段設計的基本原理和在醫療器械臨床試驗中的應用,并以某外周血管支架產品安全性和有效性評價為例,闡述其使用流程和注意事項。
基本原理
兩階段設計的本質是實現非隨機臨床試驗對隨機對照臨床試驗隨機化和前瞻性的模擬。隨機化保證了觀測到和未觀測到的變量在試驗組和對照組間的分布是均衡的,進而依據因果推斷的原理得到效應的無偏估計。前瞻性原則,即在方案設計階段研究者無法提前接觸到結局數據,使得試驗結果客觀可靠[8-9]。
1.傾向性評分與隨機化原則
非隨機臨床試驗的基線變量若未經處理,往往存在組間不均衡的情況。這些不均衡變量的維數很高且不少為連續型變量,此時觀察性研究中常用的混雜校正方法如匹配、分層、回歸并不適用:匹配能夠校正的因素非常有限;即便進行了分層,當每層的人數很少甚至沒有時,無法進行效應估計;多因素校正則要求有足夠數量的結局事件,而許多臨床試驗并不滿足條件。此時隨機化的效果可以通過傾向性評分來實現,該方法最早由Rosenbaum和Rubin于1983年提出[5],在2002年被美國FDA采納用于醫療器械上市前的非隨機臨床試驗及藥物和器械上市后的監管研究中[10]。
傾向性評分定義為在觀察到的協變量(Xi)條件下,研究對象i(i=1,2,…,n)被分配到試驗組(Zi=1)而非對照組(Zi=0)的條件概率,可表達為:
e(Xi)=P(Zi=1|Xi)
該方法基于降維的思想,當模型中納入了全部的混雜變量時,每個試驗對象的基線信息便能夠被整合至一維的傾向性評分中,可通過logistic回歸模型實現。進一步地,只需對傾向性評分進行分析,如匹配、分層、回歸或逆概率加權等,便能同時均衡兩組間全部混雜變量,實現隨機化的模擬。
2.兩階段設計與前瞻性原則
在非隨機臨床試驗中,為確保傾向性評分估計與最終的結局分析獨立進行,避免前瞻性原則遭到破壞,需要利用兩階段設計(two-stage design)[7-8]對試驗流程進行人為的劃分,如圖1所示。

圖1 兩階段設計的基本流程
兩階段設計包含兩個層次,首先試驗被嚴格地分成方案設計階段(design phase)和結局分析階段(analysis phase)。方案設計階段主要包括傾向性評分建模、模型評價和調整、制定和完善統計分析計劃等步驟,在此期間試驗正式開展,但所有結局數據(特別是當采用外部數據作為對照時)均需由防火墻隔離,在整個方案設計階段統計師都無法獲得。當數據庫鎖定,進入結局分析階段,傾向性評分模型不得更改,統計師在校正傾向性評分的基礎上對結局數據進行分析,得到最終統計分析結果。數據防火墻的建立是劃分方案設計階段與結局分析階段的關鍵,也是兩階段設計在實際應用中的最大挑戰,可通過第三方機構如數據安全監查委員會(DSMB/DMC)[11]的介入、數據分析平臺的訪問權限控制、留痕功能[12]或其他方式實現,但無論采用何種方法,都必須在制定方案之初與臨床試驗法規監管部門充分溝通達成一致。
在此基礎上,方案設計階段被進一步分為第一階段和第二階段。從開始方案設計到試驗正式開始為第一階段,主要任務包括選定獨立的統計師,確定采集的基線變量,選擇適合的對照組和估算樣本量。另外,需要在方案中明確傾向性評分模型納入變量的原則、組間平衡的評價標準和結局分析階段校正評分的方法,在方案定稿并交由CFDA備案后,這些信息便不可更改。當完成受試者入組與基線信息采集,基線數據清理完畢,第一階段即完成,進入第二階段。統計師進行傾向性評分估計,根據評分進行匹配或者分層,對組間基線變量的均衡情況進行評價。當兩組評分的重疊部分過小時,可能需要對模型進行調整和優化,直至兩組基線變量均衡可比,并最終完善統計分析計劃。研究者在方案設計階段應當與法規監管部門進行充分溝通,并于第一階段和第二階段結束后及時進行備案,缺少這些環節將嚴重影響最終試驗結果的可靠性。
實例分析
選取真實臨床試驗中的部分數據作為案例,對兩階段設計的具體流程加以闡述。某外周血管支架新一代產品的非劣效臨床試驗,假設試驗組與對照組主要終點指標術后12個月無靶血管或靶病變再次血運重建發生率均為92%,非劣效界值δ為-5%,統計檢驗的顯著性水平(α)取單側0.025,考慮脫落率10%,當每組受試者525例時,得到非劣效結論的檢驗效能(1-β)超過80%。本案例中由于試驗支架的前代產品已上市,兩代產品相似度高,適用人群一致,且前代產品的隨機對照試驗開展時間接近,積累了高質量的試驗數據,故選擇這部分數據作為本次試驗的對照。研究者對于試驗數據的全部操作均在可留痕的數據分析平臺上進行,并根據不同的角色分工和試驗的不同階段給予不同的平臺訪問權限。同時,在方案中明確了試驗需要采集的關鍵變量,入選及排除標準等信息。基于臨床經驗及既往文獻,在第一階段我們確定了12個對主要終點指標有影響的混雜變量。此外,傾向性評分模型還將納入組間有顯著差異的基線變量。傾向性評分采用1:1最鄰近法(nearest neighbor)進行匹配,當模型調整后標準化均數差小于0.1時認為變量在組間分布均衡,在結局分析階段傾向性評分通過分層進行校正。
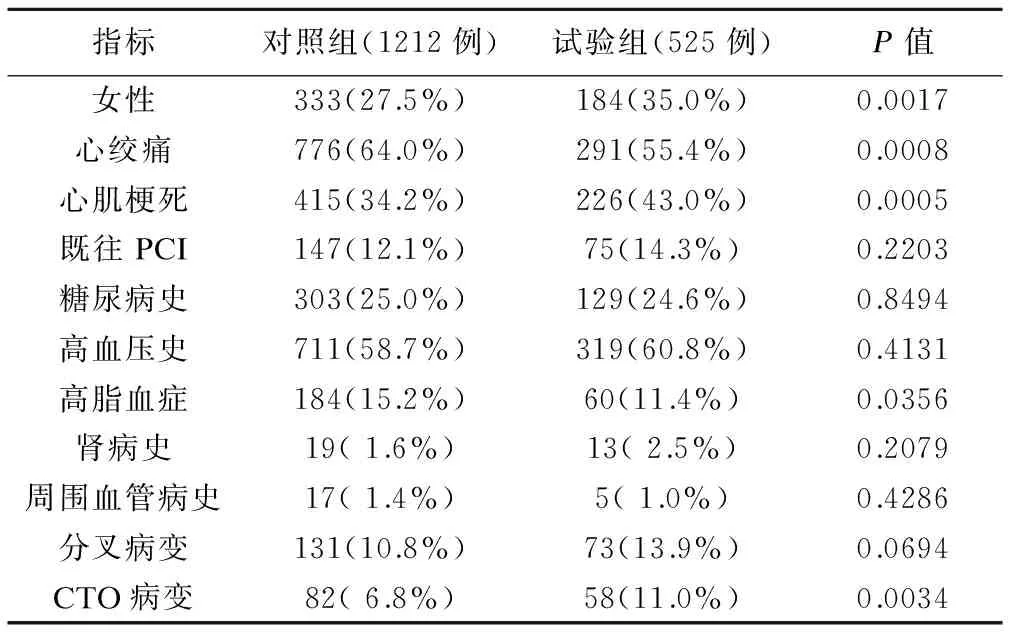
表1 試驗組和對照組納入傾向性評分模型基線定性變量比較
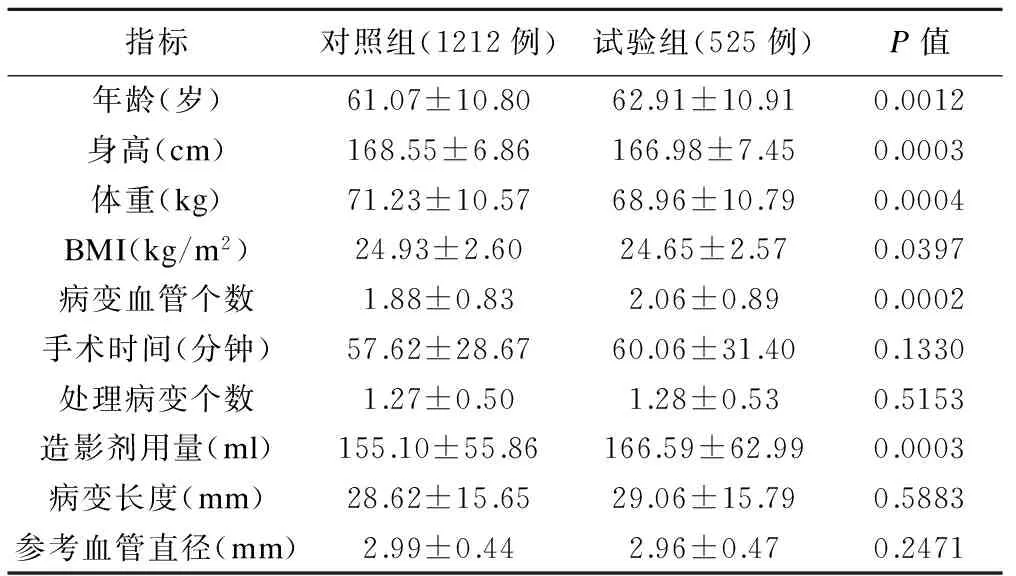
表2 試驗組和對照組納入傾向性評分模型基線定量變量比較
表1及表2展示了最終納入模型的21個基線變量在試驗組和對照組的分布情況。選擇logistic回歸模型估計兩組的傾向性評分,并對評分進行匹配,匹配后試驗組和對照組各525例。比較分析匹配后的基線變量,得到標準化均數差均小于0.1(圖2),說明匹配后的基線指標組間均衡。
在結局分析階段,對匹配后的人群進行傾向性評分分層(表3),分別在每層進行比較并通過CMH卡方計算總效應估計值,試驗組術后12個月無靶血管或靶病變再次血運重建發生率為94.48%,對照組為96.76%,率差(試驗組-對照組)為-1.76%(95%CI:-4.09%~0.63%),由于下限-4.09%大于方案中預先設定的非劣效界值-5.0%,可以認為試驗組的有效性非劣于對照組(P=0.007)。
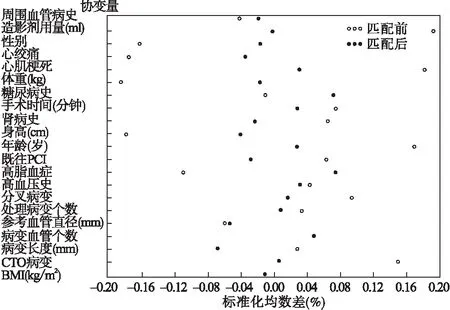
圖2 傾向性評分調整前后基線變量的標準化均數差
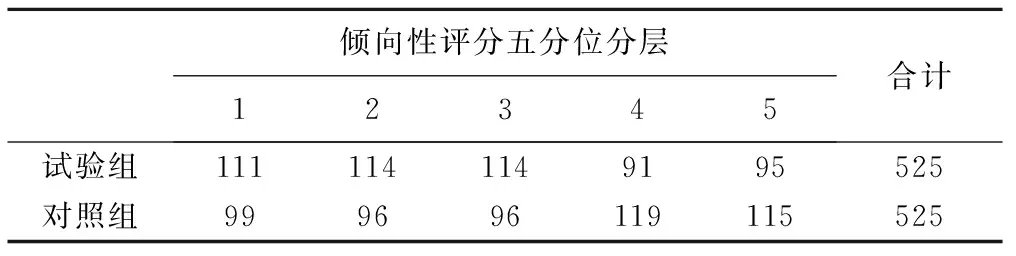
傾向性評分五分位分層12345合計試驗組1111141149195525對照組999696119115525
討 論
由于醫療器械隨機對照試驗的特殊性,在實際開展過程中往往會面臨不少倫理學和可行性問題。對于市場上已有成熟同類產品的試驗產品,當知情同意獲取困難、受試者可能不同意隨機分配等情況出現時,非隨機臨床試驗可以作為隨機對照試驗的補充,從一定層面上提供產品的安全性和有效性證據。此外,非隨機臨床試驗具有控制時間和經濟成本、充分利用高質量外部數據等優勢。兩階段設計為非隨機臨床試驗提供了嚴謹的流程規范。本文在介紹兩階段設計原理的基礎上,以實際臨床試驗數據為案例,闡述了其在醫療器械非隨機臨床試驗中的具體使用步驟。
在實際運用兩階段設計時,應當注意如下問題:
1.從監管角度而言,建立合理的防火墻機制,避免已有的結局數據發生泄漏、統計師在獲取結局數據后進行重復分析是確保非隨機臨床試驗前瞻性的關鍵。通過第三方機構如DSMB/DMC介入、數據分析平臺的訪問權限控制與留痕功能來實現前期的數據保密是可取的,此外也可以采用其他方式建立數據防火墻,但均需要在試驗方案中寫明,并于試驗正式開展前與臨床試驗法規部門進行溝通、達成一致。
2.兩階段設計對非隨機臨床試驗的流程進行了嚴格的劃分,當某階段結束后,方案中早已明確的傾向性評分模型納入變量原則、結局評價過程中評分校正方法等信息便不能更改。因此在整個試驗期間,研究者需要與臨床試驗法規部門進行多次溝通,并在每一階段結束后及時備案,否則將嚴重損害試驗的可靠性,導致最終結果不被認可。
3.對照組數據可以是已上市同類產品的同期試驗數據、同類產品的既往臨床試驗數據或注冊登記研究,其中同期試驗數據可靠性最高。若采用既往臨床試驗數據,應當注意兩組人群的入組時間應盡可能接近,入選和排除標準盡量一致,否則指南的更新、醫療實踐和技術的發展都會造成人群選擇和臨床結局定義及判斷的差異,影響數據可比性。此外,關鍵變量缺失會造成潛在的混雜效應無法處理,增加結果評價的不確定性,因此在方案設計第一階段應當明確需要采納的變量,選擇包含關鍵變量的對照組數據。
4.當兩組數據可比性不高時,在校正傾向性評分的過程中試驗樣本量會發生較大變化,檢驗功效亦會受損。因此在方案設計的第二階段,統計師應當基于當前的傾向性評分模型對樣本量和檢驗功效進行重新估計,并在統計分析報告中寫明。
5.對于隨訪周期很短或被試產品的主要評價指標為即刻終點的臨床試驗,由于較難進行明確的流程劃分,因此不建議采用傾向性評分的兩階段設計方法。
總體來說,兩階段設計能夠克服非隨機臨床試驗的主觀性問題,類似于隨機臨床試驗,并獲得客觀可靠的結論,該方法具有一定的應用價值。
[1] Yue LQ.Statistical and regulatory issues with the application of propensity score analysis to nonrandomized medical device clinical studies.Journal of Biopharmaceutical Statistics,2007,17(1):1-13.
[2] Sedgwick P.What is a non-randomised controlled trial.BMJ,2014:348.
[3] Fairman RM,Criado F,Farber M,et al.Pivotal results of the medtronic vascular talent thoracic stent graft system:the VALOR trial.Journal of vascular surgery,48(3):546-554.
[4] Troisi N,Torsello G,Donas KP,et al.Endurant stent-graft:a 2-year,single-center experience with a new commercially available device for the treatment of abdominal aortic aneurysms.Journal of Endovascular Therapy,17(3):439-448.
[5] Rosenbaum PR,Rubin DB.The central role of the propensity score in observational studies for causal effects.Biometrika,1983,70(1):41-55.
[6] Yue LQ,Lu N,Xu Y.Designing premarket observational comparative studies using existing data as controls:challenges and opportunities.Journal of biopharmaceutical statistics,2014,24(5):994-1010.
[7] Yue LQ,Campbell G,Lu N,et al.Utilizing national and international registries to enhance pre-market medical device regulatory evaluation.Journal of biopharmaceutical statistics,26(6):1136-1145.
[8] Rubin DB.For objective causal inference,design trumps analysis.The Annals of Applied Statistics,2008:808-840.
[9] Rubin DB.Using propensity scores to help design observational studies:application to the tobacco litigation.Health Services and Outcomes Research Methodology,2001,2(3):169-188.
[10]Yue LQ.Regulatory considerations in the design of comparative observational studies using propensity scores.Journal of Biopharmaceutical Statistics,2012,22(6):1272-1279.
[11]Fleming TR,DeMets DL,Roe MT,et al.Data monitoring committees:Promoting best practices to address emerging challenges.Clinical Trials,2017,14(2):115-123.
[12]Krishnankutty B,Bellary S,Kumar NB,et al.Data management in clinical research:an overview.Indian journal of pharmacology,2012,44(2):168.
Two-stageDesigninNon-randomizedMedicalDeviceClinicalTrials
Zhao Yanyan,Xu Yujun,Wang Yang,et al
(StateKeyLaboratoryofCardiovascularDisease,FuwaiHospital,NationalCenterforCardiovascularDiseases,ChineseAcademyofMedicalSciencesandPekingUnionMedicalCollege(100037),Beijing)
ObjectiveThis article introduces the concept,process as well as considerations of two-stage design.The application to non-randomized medical device clinical trials is further illustrated by example of aperipheral vascular stent study.MethodsTwo-stage design based on propensity score serves to make non-randomized clinical trials randomized and prospectively.The details of design process are presented through a non-inferiority trial evaluating the efficacy of a peripheral vascular stent.ResultsTwo-stage design handles the issue of potential biases due to non-randomization and subjective uncertainty when applying propensity score to non-randomized clinical trials,aiming to mimic randomized clinical trials and ensure the objectivity and prospectiveness of both trial design and statistical analysis.ConclusionWith great potential,two-stage design enhances the feasibility of clinical trial and makes it possible to get full use of existing data of high quality,shorten the duration of a trial,improve trial efficiency and draw a reliable conclusion ultimately.
Two-stage design; Propensity score; Medical device; Non-randomized clinical trial
1.中國醫學科學院,北京協和醫學院,國家心血管病中心,阜外心血管病醫院,心血管疾病國家重點實驗室(100037) 2.國家心血管病中心
?共同第一作者:趙延延,許毓君
△通信作者:李衛,E-mail:liwei@mrbc-nccd.com
郭海強)
猜你喜歡
河北畫報(2020年8期)2020-10-27 02:54:06
現代裝飾(2020年7期)2020-07-27 01:27:42
流行色(2020年1期)2020-04-28 11:16:38
電子制作(2019年19期)2019-11-23 08:41:36
電子制作(2019年15期)2019-08-27 01:11:50
電子制作(2019年7期)2019-04-25 13:18:16
藝術啟蒙(2018年7期)2018-08-23 09:14:18
海峽姐妹(2017年7期)2017-07-31 19:08:17
Coco薇(2017年5期)2017-06-05 08:53:16
商周刊(2017年26期)2017-04-25 08:13:04
