Au@Ag納米粒子表面增強拉曼光譜法高靈敏檢測孔雀石綠
2018-02-08 09:46:32王利華王佳慧韓艷云朱運峰吳文輝肖康飛戰藝芳曾令文
武漢工程大學學報 2018年1期
關鍵詞:檢測
王利華,王佳慧,韓艷云,朱運峰,吳文輝,肖康飛,戰藝芳,曾令文
武漢市農業科學院環境與安全研究所,湖北 武漢 430074
孔雀石綠(malachite green,MG),結構式見圖1,是一種人工合成的三苯甲烷類有機化合物,既是染料也是細菌、真菌等的優質殺菌劑[1]。因其對水產類魚蝦體水霉病、魚卵水霉病以及鰓霉病等細菌性疾病有良好的防治效果,20世紀30年代起就被廣泛用于漁業生產[2]。MG加入養殖水中后,容易被魚、蝦等吸收并代謝為無色MG,兩者均具有較高毒性和致癌性[3-4]。因此,自20世紀90年代開始,世界上許多國家相繼禁止其在可食用的水產類養殖中使用。我國于2002年也將MG列入了《食品動物禁用的獸藥及其化合物清單》,并明確禁止其用于所有食用動物的養殖[5]。盡管如此,由于MG殺菌的高效性、廉價性,且暫時沒有其他理想的替代藥物,目前一些不法漁民仍將其用于水產養殖中,這種不法行為勢必對人們的生命健康造成重大隱患。因此,在水產類產品流入市場之前,有必要進行MG檢測。
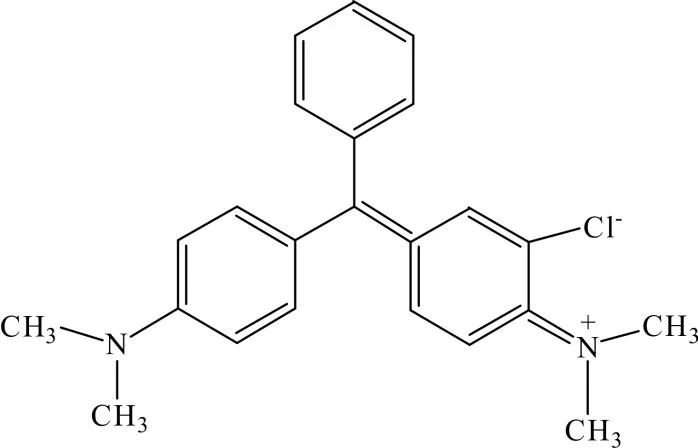
圖1 孔雀石綠鹽酸鹽的結構式Fig.1 Structure of malachite green hydrochloride
MG傳統的檢測方法主要有液相色譜法、液相色譜-質譜聯用法、氣相色譜-質譜聯用法等[6-7]。盡管這些方法靈敏度高、準確度高,但均需要大型昂貴的儀器、專業的操作人員以及復雜的樣品前處理等,檢測時間長,無法滿足水產類產品日益增長的現場快速篩查需求。表面增強拉曼光譜技術(surface enhanced Raman scattering,SERS)作為一種新興的指紋光譜式檢測技術,具有靈敏度高、特異性好、檢測快速、操作簡便且可實現無損檢測的特點,在食品安全快檢領域越來越受到重視[8-10],它是現有快檢方法中最有可能實現“傻瓜式”檢測的技術之一。近年來,基于SERS技術的MG現場檢測方法已有相關報道。例如,顧振華等[11]基于納米金和便攜式拉曼光譜儀發展了一種養殖水中孔雀石綠快速檢測方法。盡管該方法簡單、快速,但靈敏度不夠高,檢測限為5.0 μg/L。眾所周知,SERS技術最核心的內容是制備均一性好、增強效果好的金/銀納米材料充當增強基底實現待測對象的高靈敏檢測。在金/銀納米材料中,盡管納米金具有良好的穩定性,但其增強效果明顯劣于銀納米材料;而單純的銀納米材料在空氣中又易氧化[12]。因此,考慮到銀包金材料不僅具有良好的穩定性且增強效果非常好,本文通過晶種增長-抗壞血酸室溫還原法合成了Au@Ag納米材料,并以此為基底,采用SERS技術,成功構建了一種基于便攜式拉曼光譜儀的MG現場快速高靈敏檢測方法,實際可實現的檢測限低至0.5 μg/L。
1 實驗部分
1.1 儀器和試劑
紫外可見吸收光譜(ultraviolet-visible absorption spectrum,UV-vis)在 UV 分光光度計(Shimadzu,UV-2600)上采集。樣品的透射電子顯微鏡(transmission electron microscope,TEM)圖在FEI Tecnai G2 20透射電子顯微鏡(TWIN,Japan)上測定。樣品的SERS光譜在海洋光學亞洲分公司生產的便攜式拉曼光譜儀Accuman SR-510 Pro上采集,其所用的激光光源波長為(785±0.5)nm,功率350 mW。對于每一個樣品,均在基底的不同位置至少采集3個SERS光譜,然后取平均值。
氯金酸購自上海九鼎化學科技有限公司,抗壞血酸(質量分數為99%~100.5%)、檸檬酸鈉和檸檬酸(質量分數為99%)購自美國Sigma-Aldrich公司,硝酸銀購自中國上海試劑一廠,MG購自蘇州快捷康生物技術有限公司,結晶紫(crystal violet,CV)購自國藥集團化學試劑有限公司。所有其它試劑均為分析純或更高純度,且使用前未進一步處理。實驗用水為Milli-Q超純水(阻值大于18.2 MΩ·cm),基圍蝦購自武漢市白沙洲大市場。
1.2 納米金的合成
納米金的合成參照文獻[13]采用兩步增長法合成。首先,合成增長所需的晶種。典型的過程如下:在燒瓶中,加入 2.5 mL HAuCl4·3H2O(質量濃度為2 g/L)溶液,稀釋至50 mL,劇烈攪拌,加熱至沸騰后,快速加入2 mL檸檬酸鈉-檸檬酸混合溶液(其質量濃度分別為10 g/L和5 g/L)。繼續加熱攪拌5 min,冷卻至室溫備用。
第一步增長:取3 mL晶種稀釋至20 mL,室溫下劇烈攪拌,將溶液A和B同時緩慢加入燒瓶中;之后在油浴條件下加熱至沸騰,繼續攪拌30 min,最后冷卻至室溫。
第二步增長:取4.5 mL第一步增長獲得的溶液稀釋至20 mL,室溫下劇烈攪拌,將溶液A和B分別同時緩慢加入燒瓶中;之后在油浴中加熱至沸騰,繼續攪拌30 min,最后冷卻至室溫備用。
以上兩步增長中溶液A和溶液B的配制:
溶液A:2 mL HAuCl4·3H2O(質量濃度為2 g/L)溶液用水稀釋至10 mL;
溶液B:0.5 mL抗壞血酸和0.25 mL檸檬酸鈉(其質量濃度均為10 g/L)混合用水稀釋至10 mL。
1.3 Au@Ag納米顆粒的合成
Au@Ag納米顆粒的合成參照文獻[14]進行。典型的合成過程如下:在5 μL納米金溶液中依次加入200 mL檸檬酸鈉溶液(38.8 mmol/L)、50 μL抗壞血酸溶液(0.1 mol/L),室溫劇烈攪拌下緩慢加入200 μL硝酸銀溶液(0.01 mol/L),持續劇烈攪拌約15 min后,將產物保存在4oC冰箱中備用。
1.4 MG標準品以及實際樣的SERS檢測
MG標準溶液的配制:準確稱取1.0 mg MG標準品粉末溶于1 mL乙腈中,制得1 g/L標準溶液。然后用水依次稀釋不同倍數,分別配制100 μg/L、50 μg/L、10 μg/L、5 μg/L、3 μg/L、2 μg/L、1 μg/L的MG標準溶液。
MG標準品的SERS檢測:200 μL Au@Ag納米粒子溶液中,依次加入30 μL助劑H(NaCl和KBr的混合溶液),200 μL待測標準溶液,混勻后進行SERS測試。采集頻率為5 s 5次。CV標準溶液的測試與MG類似,只需將MG標準品換成CV標準品即可。
MG實際樣檢測:首先,將菜市場購買的基圍蝦蝦肉在國標的基礎上稍加改變進行相關的前處理,直接檢測時并未檢出。隨后將處理后獲得的溶液用作基質,向其中加入MG的標準溶液,配成模擬樣本進行檢測。具體測試過程與標準品類似。
2 結果與討論
2.1 納米金和Au@Ag納米粒子的表征
首先,對納米金的合成過程進行了UV-vis表征。如圖2(a)所示,初始合成的晶種顆粒在518.5 nm處有最大吸收峰,其可歸屬為納米金的表面等離子體共振(surface plasmon resonance,SPR)峰。隨著HAuCl4溶液的加入,逐步增長后,該峰依次紅移至520 nm和535 nm。對二次增長合成的金顆粒進一步進行TEM表征發現,此時的金顆粒為球形,且分散性較好、粒徑較均一,如圖2(a)中的插圖所示。由于Au@Ag納米粒子的SERS增強效果明顯優于納米金[12],以二次增長合成的金納米顆粒為種子,進一步合成了Au@Ag納米粒子。從圖2(b)中UV-vis可以看出,相對金納米顆粒而言,此時的Au@Ag納米粒子在402 nm和521 nm處有兩個最大吸收峰,其分別歸屬為Au@Ag納米粒子中銀殼和金核的SPR峰。Au@Ag鈉米粒子的TEM結果表明,因銀殼的包裹,此時粒子的粒徑相對增大,約100 nm,如圖2(b)中的插圖所示。
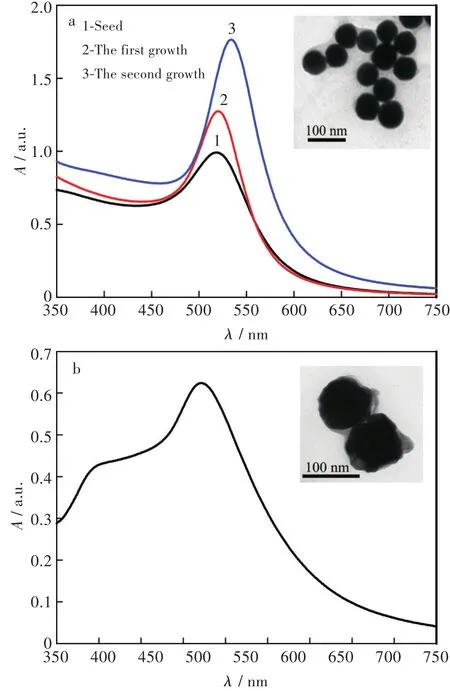
圖2 (a)納米金和(b)Au@Ag納米粒子的UV-vis和TEM圖Fig.2 UV-vis and TEM images of(a)Au nanoparticles and(b)Au@Ag nanoparticles
2.2 MG的表面增強拉曼光譜圖
用SERS技術對10 μg/L MG的可行性檢測進行了驗證。如圖3所示,Au@Ag納米粒子及Au@Ag納米顆粒-助劑H的混合溶液均沒有明顯拉曼峰。當10μg/LMG標準溶液加入后,在438cm-1、1170cm-1、1 617 cm-1處均顯示出MG的特征峰,這與文獻[15-16]報道一致,可作為MG定性檢測的依據。
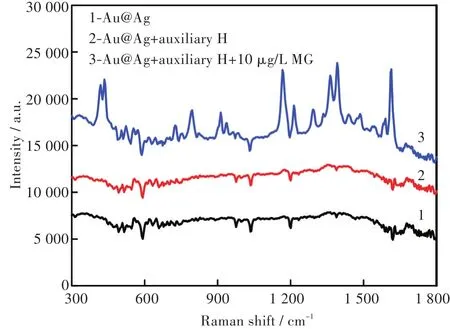
圖3 不同溶液的SERS圖Fig.3 SERS spectra of different solutions
2.3 MG檢測條件的優化
為提高MG檢測的靈敏度,本文對測試過程中Au@Ag納米粒子中銀層厚度、Au@Ag納米粒子的體積、助劑H的體積以及MG樣品的加樣順序進行了優化,該優化過程均以3 μg/L MG在1 617 cm-1處峰作為參考(見圖4中虛線框)。
由于不同銀層厚度的Au@Ag納米粒子增強效果不一樣,通過固定Au@Ag納米粒子合成過程中納米金的粒徑以及改變AgNO3加入的量,首先對Au@Ag納米粒子的銀層厚度進行了優化。如圖4(a)所示,隨著AgNO3體積的增加,Au@Ag納米粒子中銀殼逐漸變厚,SERS增強效果逐漸明顯;當AgNO3體積增加至 200 μL時,1 617 cm-1處峰強達到最大;而當AgNO3體積繼續增加時,形成的Au@Ag納米粒子溶液變得更加渾濁且容易聚沉,增強效果有下降的趨勢。因此,選取AgNO3的最佳體積為200 μL。
接著,對增強試劑的量進行了優化。如圖4(b)所示,隨著Au@Ag納米粒子體積的增加,MG在1 617 cm-1處峰強逐漸增大;當體積增加至200 μL時,峰強達到最大值;繼續增加體積,增強效果有下降的趨勢。該現象表明,Au@Ag納米粒子的體積為200 μL時,MG在其表面的單分子層吸附已達到最大值。因此,選取Au@Ag納米粒子的最佳體積為200 μL。
本文中助劑H為NaCl和KBr的混合溶液,其加入Au@Ag納米粒子溶膠中后會導致納米顆粒發生聚集,使其局域等離子電磁場發生變化,繼而在納米顆粒間產生許多“熱點”[17]。當MG分子處于這些“熱點”位置時,其拉曼信號會得到極大增強。其中,納米顆粒聚集程度的高低直接影響“熱點”的數量以及溶膠的沉降速度;而聚集過程中處于“熱點”位置的MG分子數進一步影響其拉曼信號的放大倍數,最終影響其檢測靈敏度。基于此,本文進一步對助劑H的體積進行了優化。從圖4(c)可以看出,隨著助劑H體積的增加,1 617 cm-1處峰強逐漸增大;當體積增加至30 μL時,峰強達到最大值;繼續增加體積,增強效果逐步下降,當體積增加至80 μL時,1 617 cm-1處峰幾乎消失。該現象表明,當助劑H體積為30 μL時,處于“熱點”位置的MG分子達到最大值,此時再增加助劑體積,導致Au@Ag納米顆粒聚集速度過快,很快下沉到檢測器皿底部,MG分子難以順利到達“熱點”位置,反而會影響MG拉曼信號的放大,起不到增強效果。因此,選取助劑H的最佳體積為30 μL。
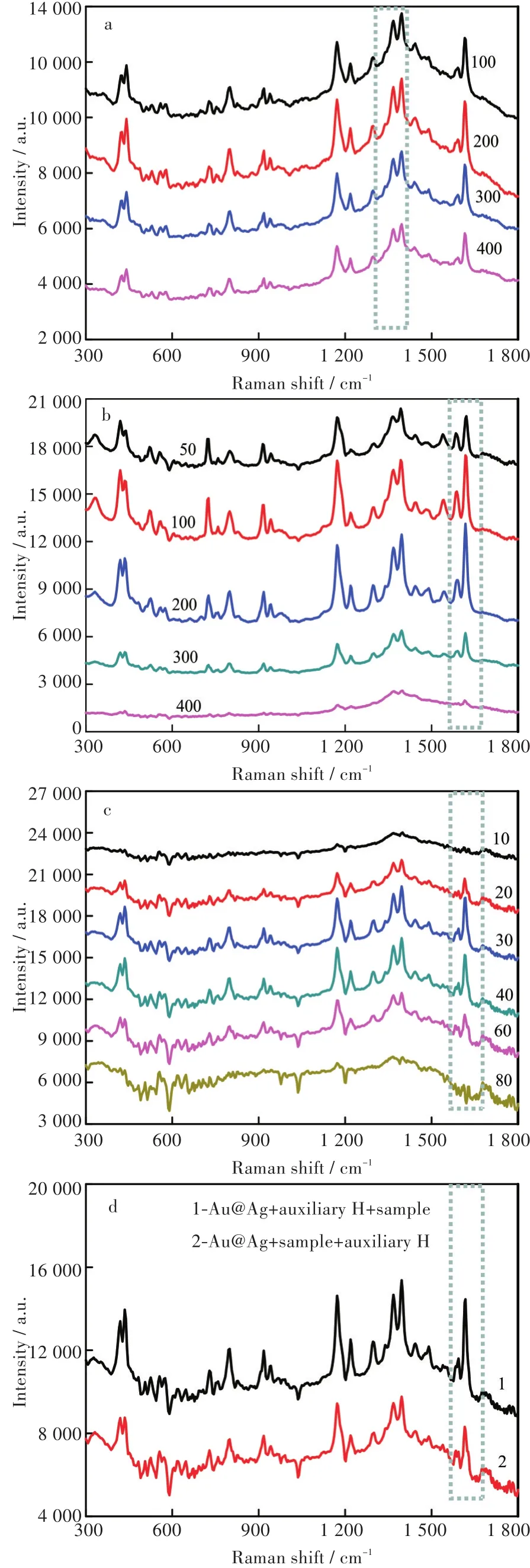
圖4 檢測條件優化:(a)不同體積AgNO3對應的Au@Ag納米粒子;(b)Au@Ag納米粒子溶液體積;(c)助劑H體積;(d)加樣順序(體積單位:mL)Fig.4 Optimized test conditions:(a)Au@Ag nanoparticles with different volumes of AgNO3;(b)Volumes of Au@Ag nanoparticles;(c)Volumes of auxiliary H;(d)Order of sample added
最后,對檢測過程中的加樣順序進行了優化。如圖4(d)所示,當待測液在助劑H之后加入時,1 617 cm-1處峰強明顯增大。這可能因為助劑中的鹵素離子優先吸附到Au@Ag納米粒子表面[18],當待測液加入時,更有利于促進MG分子吸附到Au@Ag納米粒子表面,從而使增強效果變得更強。因此,選用增強試劑、助劑H、待測液為測試時的最佳加樣順序。
2.4 靈敏度實驗
基于上述實驗獲得的優化條件,本文進一步考察了該方法檢測MG的靈敏度情況。如圖5(a)所示,隨著MG質量濃度的增加,其在1 617 cm-1處的特征峰逐漸增強。當MG質量濃度為0.5 μg/L時,其在 438 cm-1、1 170 cm-1、1 617 cm-1處的特征峰仍明顯可見。該方法最終檢測MG的線性范圍為0.5 μg/L~10 μg/L(R2=0.991),如圖 5(b)所示,實際可檢測到的最低濃度為0.5 μg/L,優于現有基于便攜式拉曼光譜儀的相關文獻報道(例如,顧振華等[11]檢測養殖水中孔雀石綠檢測限為 5 μg/L、李春穎等[19]檢測孔雀石綠標準品檢測限為0.8 μg/L),表明該方法檢測MG靈敏度很高。
此外,CV與MG具有類似的殺菌作用,常被一起用于水產養殖中[20-21]。因此,本文嘗試用該方法對CV進行測試。從圖6(a)可以看出,該方法對1 μg/L CV具有非常好的測試效果,其在802 cm-1、941 cm-1、1 170 cm-1、1 620 cm-1處的特征峰信號非常明顯[14]。
2.5 實際樣測試
將基圍蝦蝦肉進行前處理,與增強試劑Au@Ag納米粒子以及助劑H混合后,直接進行SERS檢測,未檢出MG。為測試該方法在實際樣本中的檢測能力,以蝦肉前處理獲得的溶液作為基質,配制10 μg/L和20 μg/L的MG模擬樣本溶液進行測試。如圖6(b)所示,由于提取液中非組分(如小分子蛋白質)的干擾,拉曼譜圖中熒光背景增強明顯,拉曼峰強度較同濃度標準品有明顯下降。盡管如此,模擬樣本中MG質量濃度為10 μg/L時,其特征峰仍較好分辨。該結果表明,作為一種快速定性檢測方式,該方法有望應用于實際樣品的檢測中。

圖5 (a)不同濃度MG的SERS圖;(b)工作曲線(質量濃度單位:μg/L)Fig.5 (a)SERS spectra of Au@Ag nanoparticles with different MG mass concentrations;(b)Working curve

圖6 (a)1 μg/L CV的SERS圖;(b)MG模擬樣本的SERS圖Fig.6 (a)SERS spectrum of CV with 1 μg/L;(b)SERS spectra of artificial MG samples
3 結 語
本文采用晶種增長法合成粒徑較均一、分散性良好的納米金,并以此為種子進一步合成了Au@Ag納米粒子。基于該材料優良的SERS增強效果以及便攜式拉曼光譜儀,成功構建了一種簡單、快速、高靈敏的MG的現場定性檢測方法,對MG標準品可以實現低至0.5 μg/L的檢測。進一步的實際樣測試表明,該方法有望用于魚肉、蝦肉等水產品組織樣本中MG的現場快速篩查,對保障人們舌尖上的安全具有重要的意義。
[1] 邱緒建,林洪,江潔.漁藥MG及關聯化合物檢測方法研究進展[J]. 海洋水產研究,2005,26(2):92-96.
[2] 王群,宋懌,馬兵.水產品中MG的風險評估(一)[J].中國漁業質量與標準,2011,1(2):38-43.
[3] SRIVASTAVA S,SINHA R,ROY D.Toxicological effects of malachite green [J].Aquatic Toxicology,2004,66(3):319-329.
[4] CULP S J,BLANKENSHIP L R,KUSEWITT D F,et al.Toxicity and metabolism of malachite green and leucomalachite green during short-term feeding to Fischer 344 rats and B6C3F1mice [J].Chemico-Biological Interactions,1999,122(3):153-170.
[5] 王玉堂.禁用漁藥化合物及其危害[J].中國水產,2017(1):76-80.
[6] CHEN G Y,MIAO S.HPLC determination and MS confirmation of malachite green,gentian violet,and their leuco metabolite residues in channel catfish muscle[J].Journal of Agricultural and Food Chemistry,2010,58(12):7109-7114.
[7] 李永夫,高華鵬,姚芳,等.高效液相色譜法檢測鰻魚中MG及其代謝物殘留[J].安徽農業科學,2009,37(1):31-33.
[8] HUANG J,CHEN F,ZHANG Q,et al.3D silver nanoparticles decorated zinc oxide/silicon heterostructured nanomace arrays as high-performance surface enhanced Raman scattering substrates [J].ACS Applied Material&Interfaces,2015,7(10):5725-5735.
[9] WANG P,WU L,LU Z C,et al.Gecko-inspired nanotentacle surface-enhanced Raman spectroscopy substrate for sampling and reliable detection of pesticide residues in fruits and vegetables [J]. Analytical Chemistry,2017,89(4):2424-2431.
[10] WANG J F,WU X Z,WANG C W,et al.Facile synthesis of Au-coated magnetic nanoparticles and their application in bacteria detection via a SERS method [J].ACS Applied Material&Interfaces,2016,8(31):19958-19967.
[11] 顧振華,趙宇翔,吳衛平,等.表面增強拉曼光譜法快速檢測水產品中的孔雀石綠[J].化學世界,2011,52(1):14-16,22.
[12] SHEN A G,CHEN L F,XIE W,et al.Triplex Au-Ag-C core-shell nanoparticles as a novel Raman label[J].Advanced Functional Materials,2010,20(6):969-975.
[13] ZIEGLER C,EYCHMüLLER A.Seededgrowth synthesis of uniform gold nanoparticles with diameters of 15-300 nm[J].The Journal of Physical Chemistry C,2011,115(11):4502-4506.
[14] WANG L H,SHEN A G,LI X C,et al.Inclusion of guest materials in aqueous coordination network shells spontaneously generated by reacting 2,5-dimercapto-1,3,4-thiadiazole with nanoscale metallic silver[J].RSC Advances,2014,4(65):34294-34302.
[15] ZHONG L B,YIN J,ZHENG Y M,et al.Self-assembly of Au nanoparticles on PMMA template as flexible,transparent,and highly active SERS substrates[J].Analytical Chemistry,2014,86(13):6262-6267.
[16] HUANG J,MA D Y,CHEN F,et al.Green in situ synthesis of clean 3D chestnutlike Ag/WO3-xnanostructures for highly efficient,recyclable and sensitive SERS sensing[J].ACS Applied Material&Interfaces,2017,9(8):7436-7446.
[17] ZENG Y,WANG L H,ZENG L W,et al.A label-free SERS probe for highly sensitive detection of Hg2+based on functionalized Au@Ag nanoparticles[J].Talanta,2017,162:374-379.
[18] 杜勇,方炎.鹵離子吸附競爭對羅丹明B-金膠體系NIR-SERS的影響[J]. 光電子激光,2003,14(8):881-885.
[19] 李春穎,賴克強,張源園,等.表面增強拉曼光譜檢測魚肉中禁用和限用藥物研究[J].化學學報,2013,71(2):221-226.
[20] 張志剛,冰施,陳鷺平,等.液相色譜法同時測定水產品中MG和結晶紫殘留[J].分析化學,2006,34(5):663-667.
[21] 朱程云,魏杰,董雪芳,等.改進的QuEChERS方法用于魚肉中MG、隱色MG、結晶紫和隱色結晶紫的快速檢測[J]. 色譜,2014,32(4):419-425.
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
