桑椹縮小型菌核病菌絲裂原活化蛋白激酶基因Mmk1的克隆與分析
2018-03-01 00:19:00朱志賢于翠李勇莫榮利鄧文胡興明
湖北農業科學 2018年23期
朱志賢 于翠 李勇 莫榮利 鄧文 胡興明

摘要:根據核盤菌菌核形成相關絲裂原活化蛋白激酶Smk1的氨基酸序列設計出簡并引物進行RT-PCR,克隆獲得長度為856 bp的桑椹縮小型菌核病菌絲裂原活化蛋白激酶基因Mmk1的cDNA片段。將測序結果與GenBank數據庫進行Blastx比對后,發現克隆片段所編碼的氨基酸序列與其他真菌的MAPK氨基酸序列具有較高的同源性。實時熒光定量PCR分析表明,該基因的表達量隨著菌株的生長而減少。Mmk1基因的分子克隆為進一步研究其在菌核形成中的作用奠定了基礎。
關鍵詞:桑椹縮小型菌核病菌;簡并引物;絲裂原活化蛋白激酶
中圖分類號:Q78;S888.71? ? ? ? ?文獻標識碼:A
文章編號:0439-8114(2018)23-0143-05
DOI:10.14088/j.cnki.issn0439-8114.2018.23.034? ? ? ? ? ?開放科學(資源服務)標識碼(OSID):
果桑是以產果為主、果葉兼用型桑樹的統稱,其果實桑椹具有豐富的營養和藥用價值,花青素含量極高,抗氧化功效明顯,具有促進造血細胞生長、降血糖、降血脂等藥理作用,被衛生部列為“既是食品又是藥品”名單[1,2]。桑椹除直接食用外,目前已開發出果汁飲品、桑果酒、桑果醬、桑椹膏及花青素等產品,表現出巨大的產業發展潛力和廣闊的市場前景[3,4]。然而在果桑產業發展過程中,桑椹菌核病來勢猛、發病快,發病率高達30%~90%,有些果桑園甚至絕產[5],桑椹菌核病已成為限制果桑產業發展的瓶頸問題。
桑椹菌核病又稱白果病,目前公認且常見的菌核病有桑椹肥大型菌核病、桑椹縮小型菌核病和桑椹小粒型菌核病3種,病原菌分別為桑實杯盤菌(Ciboria shiraiana)、桑椹核地杖菌(Scleromitrula shiraiana)和肉阜狀杯菌(Ciboria carunculoides)[5,6]。菌核是桑椹菌核病菌越冬的重要場所,菌核隨病椹落地,到次年3月上、中旬桑雌花開放時,土壤中的菌核萌發形成子囊盤。子囊盤上子實層著生子囊和子囊孢子,子囊孢子是病害初侵染的主要來源[7]。因此,了解菌核形成相關的分子機制可以為病害防治提供理論依據。Chen等[8]從核盤菌中克隆到一個高度保守的ERK-type絲裂原活化蛋白激酶編碼基因(ERK-type mitogen-activated protein kinases from S. sclerotiorum,smk1),該基因在菌核形成起始階段的表達量達到最高,smk1基因沉默菌株不能形成成熟的菌核,其表達受到酸性誘導和外源環磷酸腺苷的抑制,說明smk1基因可通過MAPK途徑對環境信號作出反應并調控菌核的形成。劉蒙蒙等[9]在豬苓中同樣克隆得到與smk1同源性為73%的ERK類型MAPK基因PuMAPK,實時熒光定量PCR分析結果表明豬苓菌核形成初期,菌核中的MAPK表達量隨著菌核的快速生長而減少。MAPK是細胞內的一類絲氨酸/蘇氨酸蛋白激酶,將細胞外刺激信號轉導至細胞及其核內的重要傳遞者,其中信號調節蛋白激酶(Extracellular signal regulated protein kinases,ERKs)是MAPK家族中的3個成員之一[10]。ERKs有ERK1和ERK2兩種同工酶。一般認為,ERK主要在生長因子相關刺激引起的細胞反應中起作用,主要參與細胞生長、分化和抗凋亡作用[9]。
近年來對桑椹菌核病的研究集中在病原菌鑒定,田間流行規律調查,農業、化學防治及潛在生防菌株篩選方面[7,11-13],無菌核形成相關機理的報道。本試驗根據已報道與核盤菌菌核形成相關的Smkl氨基酸保守序列設計簡并引物,克隆桑椹縮小型菌核病病菌的絲裂原活化蛋白激酶編碼基因(mitogen-activated protein kinases from Scleromitrula shiraiana,Mmk1),初步揭示其菌核形成分子機理,以期為開發高效專一性殺菌劑,切斷菌核病的病害循環提供理論依據,為病害防控提供新的思路和線索。
1? 材料與方法
1.1? 供試菌株
桑椹縮小型菌核病菌Sd1-2(GenBank accession number:MH298851)分離自宜昌市三斗坪鎮采集的病果;克隆菌株為大腸桿菌JM109(上海唯地生物技術有限公司)。
1.2? 試劑
質粒為TaKaRa公司的pMD18TM-T vector,超純RNA提取試劑盒、RNA純化反轉錄試劑盒HiFiScript Quick gDNA Removal cDNA kit、2×Es Taq Mastermix等試劑均為康為世紀公司產品,DNA回收試劑盒MiniBEST Agarose Gel DNA Extraction kit ver.4.0、熒光定量PCR試劑盒SYBR?誖Premix Ex TaqTM購自TaKaRa公司,SYBR Green I核酸染料為北京百泰克生物技術有限公司產品,瓊脂糖、蛋白胨、酵母膏等常規試劑購自上海生工公司。氨芐青霉素(Amp)、異丙基-β-D-硫代半乳糖苷(IPTG)購自北京酷來博科技有限公司,20 mg/mL X-gal購自賽國生物科技,引物合成和測序在擎科生物科技有限公司進行。
1.3? 簡并引物的設計和篩選
將核盤菌菌核形成相關的絲裂原活化蛋白激酶Smk1氨基酸序列在NCBI中進行Blastp比對,選取與其同源的代表性氨基酸序列采用ClustalX(version 1.83)對序列片段進行比對(alignment)[14],設定參數如下:多重序列比對參數:起始空位罰分(gap opening)=10,延伸空位罰分(extension opening)=0.2,轉移權系數(transition weight)=0.5,延遲發散序列(delay divergent sequences)=30%,根據需要對比對結果進行校對[8]。登錄Blockmaker,對上述序列進行Block比對,從Block results界面登錄CodeHop數據庫,進行簡并引物設計[15],利用Oligo 7.37對篩選的引物分析做進一步篩選。
1.4? RT-PCR擴增Mmk1基因片段
將Sd1-2菌株轉入鋪有滅菌玻璃紙的PDA平板中央培養,25 ℃條件下培養20 d后收集菌絲。采用超純RNA提取試劑盒按說明書步驟提取其總RNA,用微量紫外分光光度計檢測合格后,用RNA 純化反轉錄試劑盒HiFiScript Quick gDNA Removal cDNA kit去基因組DNA和反轉錄。以反轉錄產物為模板,用設計的簡并引物DMMK1-F/ DMMK1-R擴增Mmk1的cDNA片段,反應在50 μL PCR體系中進行,包括2×Es Taq Mastermix 25 μL,10 μmol/L引物各2 μL,模板cDNA 10 ng 1 μL,加入ddH2O至體系體積達50 μL。PCR擴增參數為:95 ℃ 4 min;94 ℃ 1 min,54 ℃ 1 min,72 ℃ 1 min,35個循環;72 ℃ 10 min。
1.5? RT-PCR產物的檢測、克隆與測序
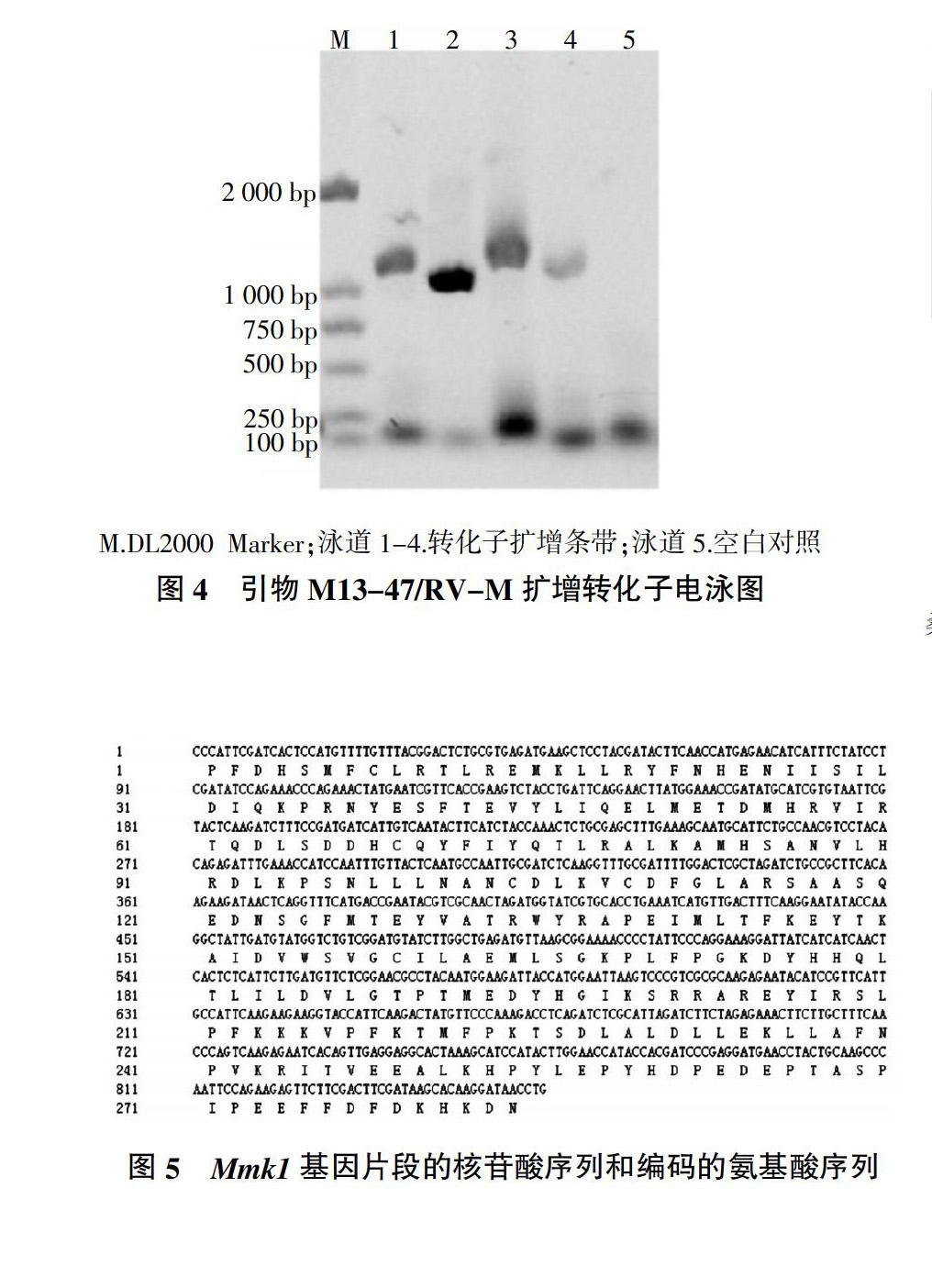
PCR產物進行1%瓊脂糖凝膠電泳和紫外觀察,切取目的條帶,用TaKaRa回收試劑盒回收,回收產物與pMD18TM-T vector進行連接后轉化JM109大腸桿菌,然后在含有AMP、IPTG和X-gal的抗性LB固體培養基平板上37 ℃培養過夜,經藍白斑篩選,挑取白色菌落,進行菌落PCR驗證,然后將陽性克隆送樣至擎科生物公司,采用載體通用引物M13-47/RV-M雙向測序分析。
1.6? Mmk1基因片段測序結果分析
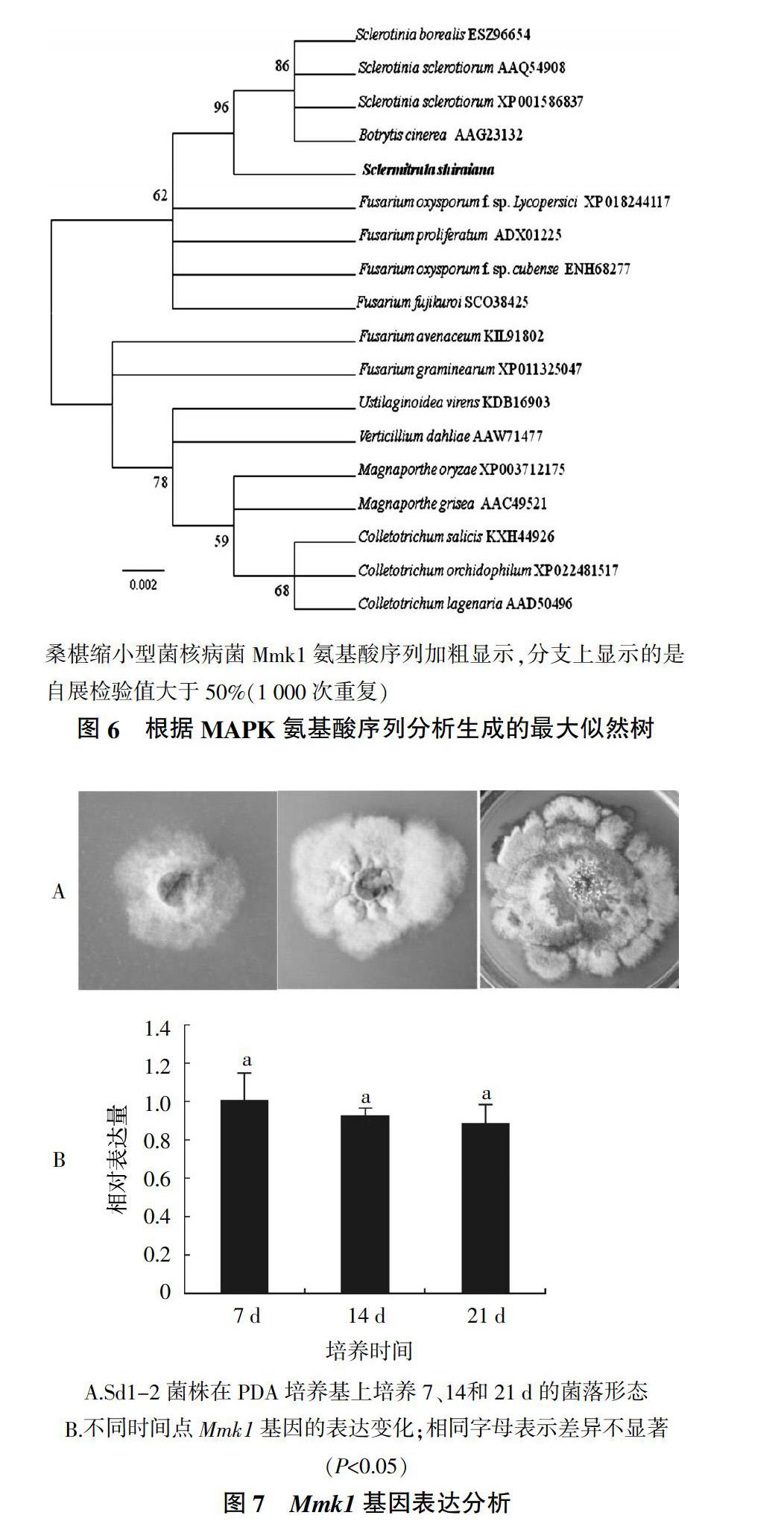
測序后的片段在NCBI中進行Blastx比對,選取與其同源的代表性氨基酸序列用ClustalX(version 1.83)進行多序列比對,用MEGA 5.05軟件中的最大似然法構建系統進化樹,選用Jone-Taylor-Thomton(JTT)模型,進化樹用自展分析法進行檢驗,循環1 000次。
1.7? Mmk1基因表達分析
為了分析Mmk1基因在不同時間點的表達情況,提取了在PDA平板上培養7、14和21 d的菌株Sd1-2菌絲RNA。反轉錄成cDNA后,根據克隆的Mmk1基因序列設計引物MMK1-F/MMK1-R:5′-ATCGTTCACCGAAGTCTACCTG-3′/5′-CTCTGTGT
AGGACGTTGGCAG-3′進行實時熒光定量PCR擴增。PCR反應設定為10 μL總反應體系,其中包括5 μL 2×SYBR?誖Premix ExTaqTM Master Mix,0.5 μL引物(2.5 μmol/L),0.2 μL ROX Reference Dye,1 μL cDNA模板和2.8 μL ddH2O。反應循環參數設定如下:95 ℃ 1 min;95 ℃ 15 s,58 ℃ 20 s,72 ℃ 45 s,40個循環。反應結束后采用2-ΔΔCt法進行計算不同處理組的相對表達量,以β-tubulin基因(5′-TTGGA
TTTGCTCCTTTGACCAG-3′/5′-AGCGGCCATCATGT
TCTTAGG-3′)作為內參基因,每個反轉錄樣品重復3次試驗。采用DPS(version 3.01)軟件對數據進行方差分析,并用LSD法比較不同處理間的差異顯著性(P<0.05)。
2? 結果與分析
2.1? 簡并引物的設計和篩選
將核盤菌菌核形成相關的絲裂原活化蛋白激酶Smk1氨基酸序列在NCBI中進行Blastp比對,選取與其同源的代表性菌株的MAPK氨基酸序列用ClustalX進行多序列比對(圖1),根據需要對比對結果進行校對,登錄Blockmaker進行Block比對,得到9個高度保守的連續氨基酸區域(Blocks),從Block results界面登錄CodeHop數據庫,進行簡并引物搜索,根據上、下游引物之間的Tm值和簡并度不能相差太大,簡并度控制在32以下;同時保證引物克隆足夠長的片段的原則。篩選出保守結構域PFDHSMFCL和FFDFDKNKDNLT設計的上、下游引物DMMK1-F/DMMK1-R:5′-CCCATTCGATCAC
TCCatgttytgyyt-3′/5′-CAGGTTATCCTTGTGCTTATC Gaartyraaraa-3′,用Oligo 7.37對其分析未發現引物二聚體和發夾結構。
2.2? RT-PCR擴增及擴增產物的克隆、測序
提取菌株Sd1-2的RNA(圖2),反轉錄合成cDNA后,用DMMK1-F/DMMK1-R為引物進行PCR擴增,PCR產物瓊脂糖凝膠電泳結果如圖3,片段大小與預測結果大小基本一致。由瓊脂糖電泳檢測結果可見,這種引物的特異性較好,基本沒有非特異條帶。切膠后,用DNA回收試劑盒回收目標片段,連接載體轉化后,用通用引物M13-47/RV-M對轉化子進行菌落PCR擴增驗證(圖4),將陽性克隆送樣測序。
2.3? Mmk1基因片段測序結果分析
利用設計的簡并引物進行RT-PCR,克隆到長度為856 bp cDNA片段(圖5)。測序后的片段在NCBI中進行Blastx比對,發現其與Sclerotinia sclerotiorum的MAPK氨基酸的同源性為99%,并都與已發表的其他真菌MAPK氨基酸高度同源。選取與其同源的代表性氨基酸序列用ClustalX(version 1.83)進行多序列比對,用MEGA 5.05軟件構建的系統發育樹結果表明Mmk1序列與Sclerotinia sclerotiorum和Botrytis cinerea的MAPK氨基酸序列聚為一類(圖6)。
2.4? Mmk1基因表達的實時熒光定量PCR檢測
利用實時熒光定量PCR檢測了在PDA平板上培養7、14和21 d Sd1-2菌株(圖7A)Mmk1基因的表達量,發現Mmk1基因的相對表達量隨著菌株培養時間的延長而減少,但差異不顯著(圖7B)。
3? 小結與討論
本研究利用CodeHop設計的簡并引物DMMK1-F/DMMK1-R,經RT-PCR擴增出特異片段,克隆獲得856 bp桑椹核地杖菌絲裂原活化蛋白激酶基因Mmk1的cDNA片段,Blastx比對發現Mmk1與核盤菌菌絲裂原活化蛋白激酶Smk1同源性為99%,系統進化樹分析發現MMK1序列與Sclerotinia sclerotiorum和Botrytis cinerea的MAPK氨基酸序列聚為一類。實時熒光定量PCR結果表明該基因的表達隨著菌株的生長而減少。
由于S. shiraiana基因組序列未知,試圖用其他已經發表的核盤菌Smk1引物[8,16]來擴增S. shiraiana中的Mmk1基因,但均未成功。經過Blastn比對發現不同物種間MAPK基因核苷酸序列差異較大,而氨基酸序列相對保守。由于密碼子偏好性差異,不同的核酸序列可能表達出相同的氨基酸序列,而氨基酸序列才具有真正的保守性,因此采用氨基酸序列設計簡并引物。在CodeHop引物的簡并設計過程中,引物搜索后得到的結果中往往會出現大量的引物序列,這時合適的篩選方法和篩選工具尤為重要[17]。本試驗首先根據目標引物位置、片段長度、引物Tm值和簡并度對上、下游引物進行初次篩選,接著利用生物軟件Oligo7.37對初次篩選結果進行分析,這樣就能夠得到較優化的引物對,使得試驗成功更有保證。
將克隆的Mmk1基因編碼的氨基酸序列與其他真菌MAPK氨基酸序列進行聚類分析,發現Mmk1序列雖然與核盤菌、灰霉菌的MAPK氨基酸序列聚為一類,但是在不同分支上,核盤菌和灰霉菌卻在同一分支上。該結果與它們的分類地位一致,桑椹核地杖菌屬于地舌菌科核地杖菌屬,而灰霉屬于核盤菌科葡萄孢盤菌屬[18],說明桑椹核地杖菌與核盤菌親緣關系較遠。該現象與蘇正川[11]報道類似,但王愛印[19]發現桑椹核地杖菌菌株SXSG-5在PDA培養基上培養9周,可在培養基內部產生黑色塊狀菌核,在查氏培養基上培養12周,亦可在其內部產生黑色顆粒狀菌核。鑒于本試驗未在PDA上發現菌株Sd1-2產生菌核,菌株在PDA平板上7 d左右才能看到有明顯的菌絲形成,14 d左右菌絲隨著菌株生長逐漸凝結形成凸起,21 d左右菌落上產生乳白色液體,鏡檢發現為該菌的分生孢子堆。這3個時間點菌株形態有較大變化,因此選擇這3個時間點檢測Mmk1基因的表達情況。核盤菌smk1和豬苓PuMAPK基因均是在菌核形成初期表達量最高,隨著菌核的生長而減少[8,9];Mmk1基因表達也是在菌絲凝結后逐漸減少,與核盤菌smk1和豬苓PuMAPK基因表達類似。本研究成功克隆了Mmk1基因部分cDNA片段,為下一步利用RACE等技術克隆該基因全長,通過RNAi等手段進一步研究該基因在桑椹縮小型菌核病菌菌核生長發育中的生物學功能提供了理論依據。
參考文獻:
[1] 操紅纓.桑椹研究進展[J].時珍國醫國藥,1999,10(8):626-628.
[2] KIM S B,CHANG B Y,JO Y H,et al. Macrophage activating activity of pyrrole alkaloids from Morus alba fruits[J].Journal of Ethnopharmacology,2013,145(1):393-396.
[3] 李玉峰.湖州市桑果產業的現狀與發展對策研究[D].杭州:浙江大學,2012.
[4] 鄒宇曉,廖森泰,肖更生,等.蠶桑資源多元化開發利用新技術研究進展[J].蠶業科學,2016(4):561-569.
[5] 蒯元璋,吳福安.桑椹菌核病病原及病害防治技術綜述[J].蠶業科學,2012,38(6):1099-1104.
[6] HONG S K,WAN G K,SUNG G B,et al. Identification and distribution of two fungal species causing sclerotial disease on mulberry fruits in korea[J].Mycobiology,2007,35(2):87-90.
[7] 呂蕊花,趙愛春,余? 建,等.桑椹肥大性菌核病病原菌生物學特性及流行性[J].微生物學報,2017,57(3):388-398.
[8] CHEN C B,HAREL A R,YARDEN O,et al. MAPK regulation of sclerotial development in Sclerotinia sclerotiorum is linked with pH and cAMP sensing[J].Mol Plant Microbe Interact,2004, 17(4):404.
[9] 劉蒙蒙,宋? 超,邢詠梅,等.豬苓菌絲形成菌核的MAPK基因克隆及表達分析[J].微生物學通報,2015,42(12):2345-2350.
[10] GOUGH N R. Focus issue:Recruiting players for a game of ERK[J].Science Signaling,2011,4(196):eg9.
[11] 蘇正川.桑葚縮小型菌核病病原學及其分子檢測體系[D].重慶:西南大學,2015.
[12] 鄭章云,楊? 義,任杰群,等.2種木霉菌制劑對桑椹菌核病的田間防治試驗[J].蠶業科學,2016(1):168-170.
[13] SULTANA R,KIM K. Bacillus thuringiensis C25 suppresses popcorn disease caused by Ciboria shiraiana in mulberry (Morus australis L.)[J].Biocontrol Science and Technology,2016, 26(2):145-162.
[14] THOMPSON J D,GIBSON T J,PLEWNIAK F,et al. The CLUSTAL_X windows interface:Flexible strategies for multiple sequence alignment aided by quality analysis tools[J].Nucleic Acids Research,1997,25(25):4876-4882.
[15] 夏? 瑞,陸旺金,李建國,等.簡并引物的程序化設計與荔枝HMGR基因片段的克隆[J].果樹學報,2006,23(6):903-906.
[16] 劉? 榮.核盤菌兩個新型RNA病毒基因組的分子生物學特性研究[D].武漢:華中農業大學,2015.
[17] 李運合,孫光明.利用CODEHOP和iCODEHOP設計簡并引物克隆芒果LAX基因家族片段[J].熱帶作物學報,2011,32(12):2278-2282.
[18] 魏景超.真菌鑒定手冊[M].上海:上海科學技術出版社,1979.
[19] 王愛印.桑椹菌核病病原菌的分離、鑒定及其拮抗性桑樹內生菌的研究[D].重慶:西南大學,2016.
