Box-Behnken法優選川烏、草烏、馬錢子超聲提取工藝△
2018-03-09 05:36:44汪坤趙旭王楠斐胡昌江
中國現代中藥 2018年1期
汪坤,趙旭,王楠斐,胡昌江
(1.成都中醫藥大學,四川 成都 611137;2.河南省中醫院,河南 鄭州 450002)
現代藥理研究表明,川烏[1]、草烏[2]、馬錢子[3]均具有抗炎、鎮痛和抗腫瘤作用,為中醫臨床治療癌癥和緩解癌性疼痛的常用藥物。河南省中醫院腫瘤科以川烏、草烏、馬錢子為主藥研制的消積止痛膏緩解癌性疼痛效果顯著,但原制劑為黑膏藥,現擬把飲片進行提取之后制成巴布劑。
文獻報道中有同時提取川烏、草烏,單獨提取馬錢子的工藝研究,而尚未有對三者同時提取的研究。本研究擬同時提取3種飲片,以烏頭堿、士的寧、馬錢子堿提取量的綜合評分為指標,優選提取工藝,為同時包含川烏、草烏、馬錢子的中藥制劑開發提供理論基礎。
川烏、草烏的藥效成分為烏頭堿等雙酯型生物堿類成分,該類成分易水解,當溶劑的pH值、溫度升高可以加速其水解過程[4-5];馬錢子的藥效成分為士的寧、馬錢子堿等生物堿類成分[5]。本實驗采用超聲提取法,以酸水為溶媒,在低溫、低pH值的條件下優化川烏、草烏、馬錢子的提取工藝。
1 儀器與試藥
1.1 儀器
Aglient-1260高效液相色譜儀(二極管陣列檢測器);AE240型十萬分之一電子分析天平(瑞士梅特勒-托利多公司);DHG-9076A型電熱恒溫鼓風干燥箱(上海精宏實驗設備有限公司);電子恒溫水浴鍋(北京中興偉業儀器有限公司);PL-S60超聲波清洗機(東莞康士潔超聲波科技有限公司)。
1.2 試藥
烏頭堿對照品、士的寧對照品、馬錢子堿對照品(四川維克奇生物科技有限公司,批號分別為110718-201108、110718-201108、110718-201108,純度均≥98%);乙腈、甲醇為色譜純;水為娃哈哈飲用純凈水。
川烏、草烏、馬錢子購于亳州藥材市場,經河南省中醫院黃小敏副主任中藥師鑒定分別為毛茛科植物烏頭AconitumcarmichaeliiDebx.的干燥母根、毛茛科植物北烏頭AconitumkusnezoffiiReichb.的干燥塊根、馬錢科植物馬錢Strychnosnux-vomicaL.的干燥成熟種子。
2 方法
2.1 色譜條件
AgilentTC-C18(2)(250 mm×4.6 mm,5 μm);流動相:乙腈(A)-0.1%磷酸二氫鈉(B);采用梯度洗脫(0~10 min,12%A;10~11 min,12%A→36%A;11~18 min,36%A;18~20 min,36%A→12%A);檢測波長分別為0~8 min時260 nm(測定士的寧、馬錢子堿),8~20 min時235 nm(測定烏頭堿);柱溫25 ℃,流速1 mL·min-1。
2.2 混合對照品溶液的制備
精密稱烏頭堿、士的寧、馬錢子堿對照品,色譜甲醇定容置于容量瓶中,質量濃度分別為0.2、0.0168、0.05 mg·mL-1。
2.3 供試品溶液的制備
稱取草烏、川烏各3 g,馬錢子1 g,按照正交試驗設計的條件進行超聲提取,提取液氨水堿化后過濾,濾液三氯甲烷萃取后收集三氯甲烷層,定容于50 mL容量瓶中,0.45 μm微孔濾膜過濾,備用。
2.4 陰性樣品溶液的制備
分別稱取川烏、草烏,馬錢子,按照2.3項下方法制備缺川烏、草烏,缺馬錢子陰性樣品溶液。
2.5 專屬性試驗
取混合對照品溶液、樣品溶液及陰性對照品溶液,按上述色譜條件分別進樣分析,士的寧、馬錢子堿、烏頭堿峰形對稱,無雜質干擾且基線平穩,保留時間分別為6.72、7.49、14.69 min,見圖1,方法專屬性良好。

注:A.混合對照品;B.樣品;C.缺馬錢子陰性樣品;D.缺川烏、草烏陰性樣品;1.士的寧;2.馬錢子堿;3.烏頭堿。圖1 混合對照品、樣品、陰性樣品HPLC圖
2.6 標準曲線的制備
分別精密移取2.2項下混合對照品溶液0.6、1.0、1.2、1.8、2.0 mL于10 mL容量瓶中,用色譜甲醇定容至刻度,制得不同濃度的混合對照品溶液,按照2.1項下色譜條件進行測定,計算峰面積值,并以混合對照品溶液濃度(X)對峰面積值(Y)回歸,得回歸方程見表1。

表1 線性關系
2.7 精密度試驗
取對照品溶液重復進樣6次,每次10 μL,按2.1項下色譜條件測定,計算烏頭堿、士的寧、馬錢子堿峰面積的RSD分別為1.35%、1.11%、1.08%,表明儀器精密度良好。
2.8 穩定性試驗
稱取草烏、川烏各3 g,馬錢子1 g,按照供試品溶液的制備方法制備供試品溶液,分別放置0、2、4、6、8、10 h后進樣,進樣量10 μL,按2.1項下色譜條件測定,計算烏頭堿、士的寧、馬錢子含量的RSD值分別為1.68%、2.37%、2.54%,表明供試品溶液在10 h內穩定。
2.9 重復性試驗
稱取樣品6份,其中草烏、川烏各3 g,馬錢子1 g,按照供試品溶液的制備方法制備供試品溶液,分別進樣10 μL,按2.1項下色譜條件測定,計算烏頭堿、士的寧、馬錢子含量的RSD值分別為2.22%、1.97%、1.78%,表明該方法重復性良好。
2.10 加樣回收率試驗
取已知烏頭堿、士的寧、馬錢子含量的樣品6份,分別加入一定量烏頭堿、士的寧、馬錢子堿對照品溶液,按照供試品溶液制備方法制備樣品溶液,分別進樣10 μL,按照2.1項下色譜條件條件測定,計算平均加樣回收率分別為2.17%、2.26%、1.98%。
3 實驗結果
3.1 單因素試驗
3.1.1 超聲功率 固定鹽酸水濃度為1.5%,加液量10倍,提取時間1 h,稱取樣品5份,草烏、川烏各3 g,馬錢子1 g。按照供試品溶液的制備方法分別在超聲功率200、300、400、500、600 W下提取制備樣品溶液,分別進樣10 μL,按2.1.1項下色譜條件測定,計算烏頭堿、士的寧、馬錢子含量,結果見表2,發現超聲功率600 W時提取效率最佳。

表2 超聲功率對川烏、草烏、馬錢子提取的影響
3.1.2 酸濃度 固定提取時間1 h,加液量10倍,稱取樣品5份,草烏、川烏各3 g,馬錢子1 g。分別加入0.5%、1%、1.5%、2%、2.5%的鹽酸水溶液,超聲功率600 W,按照供試品溶液的制備方法制備樣品溶液。分別進樣10 μL,按2.1.1項下色譜條件測定,計算烏頭堿、士的寧、馬錢子含量,結果見表3。選擇鹽酸水濃度1%、1.5%、2%為星點設計-響應面法試驗的水平條件。

表3 酸濃度對川烏、草烏、馬錢子提取的影響
3.1.3 提取時間 固定鹽酸水濃度為1.5%,加液量10倍,稱取樣品5份,草烏、川烏各3 g,馬錢子1 g,超聲功率600 W,按照供試品溶液的制備方法分別提取0.5、1、1.5、2、2.5 h,制備樣品溶液,分別進樣10 μL,按2.1.1項下色譜條件測定,計算烏頭堿、士的寧、馬錢子含量,結果見表4,選擇提取時間1、1.5、2 h為星點設計-響應面法試驗的水平條件。

表4 提取時間對川烏、草烏、馬錢子提取的影響
3.1.4 加液量 固定鹽酸水濃度為1.5%,提取時間1 h,稱取樣品5份,草烏、川烏各3 g,馬錢子1 g,分別加入6、8、10、12、15倍量的鹽酸水溶液,超聲功率600 W,按照供試品溶液的制備方法制備樣品溶液,分別進樣10 μL,按2.1.1項下色譜條件測定,計算烏頭堿、士的寧、馬錢子含量,結果見表5,選擇加液量8、10、12倍為星點設計-響應面法實驗的水平條件。

表5 加液量對川烏、草烏、馬錢子提取的影響
3.1.5 提取次數 固定鹽酸水濃度為1.5%,加液量10倍,提取時間1 h,稱取樣品5份,其中草烏、川烏各3 g,馬錢子1 g,按照供試品溶液的制備方法分別提取1、2、3次,制備樣品溶液,分別進樣10 μL,按2.1.1項下色譜條件測定,計算烏頭堿、士的寧、馬錢子含量,提取兩次后烏頭堿、士的寧、馬錢子的提取率達到90%以上,為了降低成本、提高生產效率,因此選擇提取2次。
3.2 星點設計-響應面法優化川烏、草烏、馬錢子超聲提取工藝
3.2.1 響應面試驗設計 在單因素試驗的基礎上,以酸水濃度(A)、提取時間(B)、加液量(C)為因變量,烏頭堿、士的寧、馬錢子提取量的綜合評分為相應值設計三因素三水平中心復合試驗,見表6。

表6 Box-Behnken試驗因素水平表
3.2.2 響應面試驗結果及方差分析 根據Box-Behnken法設計方案及2.1.3項下供試品溶液的制備方法進行實驗,測定烏頭堿、士的寧、馬錢子堿的含量,計算綜合評分,響應試驗結果見表7。采用Design Expert 8.06軟件對所得數據進行回歸分析,方差分析結果見表8。
表7數據經Design Expert 8.0.6軟件回歸分析,并進行擬合,得到以上3個因素對提取工藝綜合評分影響的回歸模型方程:Y=0.222 7+0.508 85A+0.418 35B+0.011 625C-0.064AB-2.75×10-3AC-3.75×10-3BC+3.750×10-3-0.133A2-0.127 2B2-5.125×10-4C2,方差分析結果見表8所示。
由表8可知,B、A2、B2對綜合評分的的影響極顯著(P<0.01),A、AB對綜合評分的的影響較顯著(P<0.05)。本實驗模型回歸P值為<0.01,說明模型回歸顯著可靠;失擬誤差P值為0.027 7>0.05,說明失擬度不明顯;說明該方程擬合度較高,實驗誤差較小,該模型可以有效地對川烏、草烏、馬錢子提取工藝進行分析和預測。
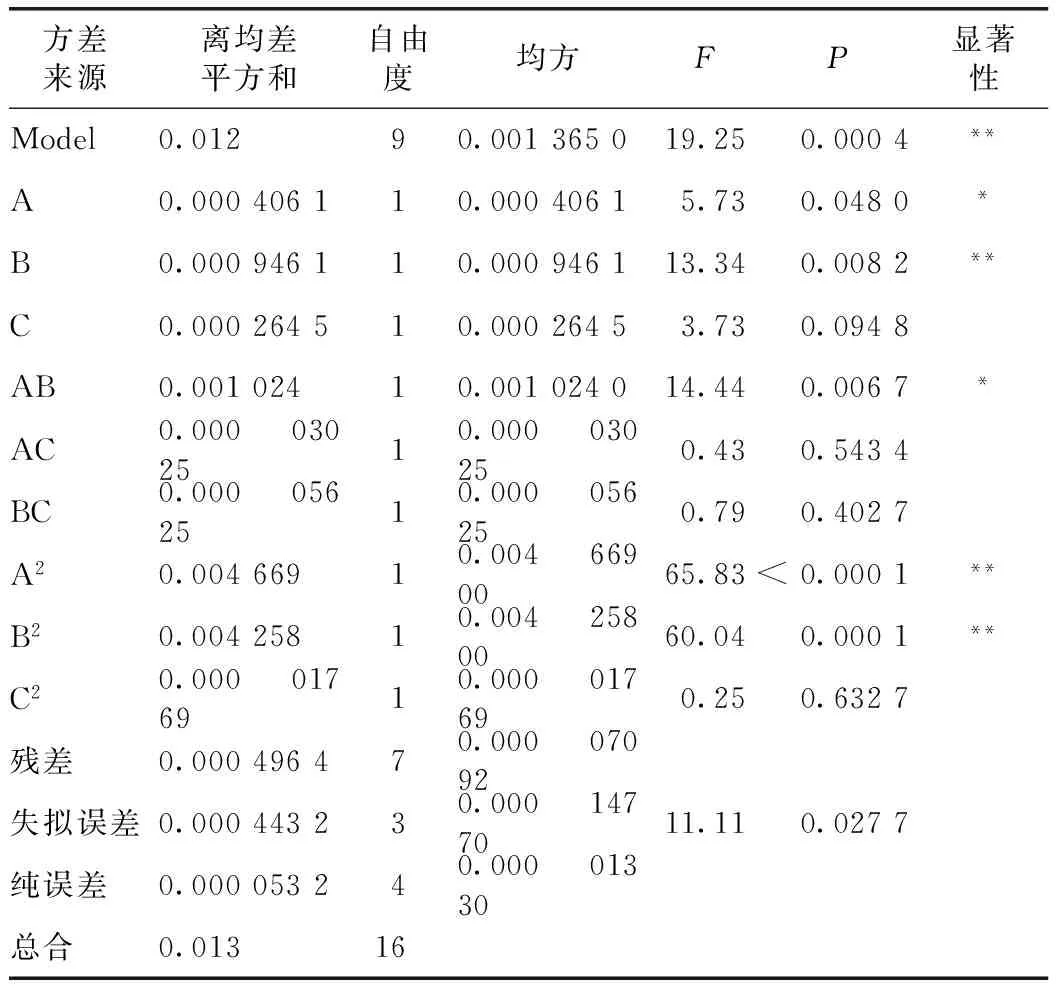
表8 回歸系數的顯著性檢驗
注:*P<0.05為顯著水平,**P<0.01為極顯著水平。
3.2.3 響應面分析 經 Design Expert 8.0.6軟件回歸分析,分別繪制各指標與響應值的響應曲面圖,用來確定各因素對川烏、草烏、馬錢子提取工藝綜合評分的影響,結果見圖2~4。由圖可知,提取時間一定時,綜合評價結果隨酸濃度的增加先升高后降低;加液量一定時,綜合評價結果隨提取時間的增加先升高后降低;酸濃度一定時,綜合評價結果隨超聲功率的增加先升高后趨于平穩。

圖2 提取時間與酸濃度交互作用的響應面圖

圖3 加液量與酸濃度交互作用的響應面圖

圖4 加液量與提取時間交互作用的響應面圖
3.2.4 工藝驗證 經 Design Expert 8.0.6軟件得到的回歸方程,求解得到草烏、川烏、馬錢子最佳超聲提取工藝:以1.41%的鹽酸水為溶媒,超聲功率600 W,11.99倍加液量提取2次,每次1.46 h,此時綜合評分的預測值為0.968。根據實際生產情況修正最佳提取工藝:1.4%的鹽酸水為溶媒,超聲功率600 W,12倍量的加液量提取2次,每次1.5 h。
按修正工藝進行3批驗證,結果見表9,綜合評分平均值為0.961,RSD值為0.82%。結果表明,建立的綜合評分與酸濃度、加液量和提取時間之間關系的回歸模型是科學合理的。

表9 驗證實驗結果
4 討論
川烏、草烏所含的藥效成分烏頭堿易水解,隨著溫度的升高,水解速度加快。目前對川烏、草烏常用的提取方法有冷浸法[7]、超聲提取法、滲漉法、回流提取法等,其中回流提取法烏頭堿水解速度較快;而冷浸法、滲漉法所用溶劑較多,用時較長,因此本實驗采用超聲波提取法,提取效率較高。
馬錢子的提取工藝研究中采用的溶劑有水、酸水、乙醇等,酸水提取效率略低于乙醇提取,但二者之間無顯著差異[8]。
本實驗采用超聲提取法用酸水提取川烏、草烏、馬錢子中的生物堿類成分,浸泡時間較短,提取液同回流提取法相比較為粘稠,但采用減壓過濾法可以快速過濾,優選的工藝簡便易行,可以為制劑生產提供參考。
[1] 劉瑤,焦豪妍.川烏毒理與藥理現代研究進展[J].云南中醫中藥雜志,2010,31(3):66-67.
[2] 白長明,巴圖德力根.草烏藥理毒理現代研究及蒙醫臨床應用現狀[J].遼寧中醫雜志,2008,35(3):475-477.
[3] 王曉崴,龔千鋒.馬錢子的炮制沿革、藥理作用及安全性的研究進展[J].江西中醫藥,2013,3(44):70-72.
[4] 隨志剛,陳明玉,劉志強,等.草烏中烏頭類生物堿提取方法比較研究[J].時珍國醫國藥,2009,20(3):513-514.
[5] 黃勤安,張聿梅,何軼,等.烏頭堿水解轉化規律的研究[J].中國中藥雜志,2007,32(20):2143.
[6] 蔡皓,王丹丹,劉曉,等.馬錢子堿、馬錢子總生物堿與馬錢子粉在大鼠體內藥動學的比較[J].中國中藥雜志,2012,37(14):2160-2163.
[7] 劉學湘,陳建偉.川烏中總生物堿提取工藝優化研究[J].中華中醫藥雜志,2009,24(1):52-53.
[8] 湯淮波,張令君,李湘玲,等.不同溶劑提取馬錢子中士的寧與馬錢子堿的實驗研究[J].中醫藥導報,2009,15(11):52-53.
