仿制藥一致性評價中溶出度試驗方法的探討
2018-03-11 20:54:12李帥龔前飛廖萍張景辰陳桂良
上海醫藥 2018年3期
李帥+龔前飛+廖萍+張景辰+陳桂良

摘 要 通過分析仿制藥一致性評價中采用的溶出度試驗方法的現狀,比較美國藥典和中國藥典中的溶出度試驗方法,重點介紹美國藥典中的溶出度試驗方法的開發與驗證,以期為仿制藥一致性評價提供適宜的溶出度試驗方法。參照美國藥典相關要求構建溶出度試驗方法可有效提高仿制藥一致性評價的合規性和效率。
關鍵詞 溶出度 美國藥典 中國藥典 一致性評價
中圖分類號:R951 文獻標識碼:C 文章編號:1006-1533(2018)03-0024-05
The discussion of dissolution test methods in consistency evaluation for generics
LI Shuai*, GONG Qianfei, LIAO Ping, ZHANG Jingchen, CHEN Guiliang**(Shanghai Center for Drug Evaluation and Inspection, Shanghai 201203, China)
ABSTRACT The dissolution test methods between Chinese Pharmacopoeia and United States Pharmacopoeia (USP) were compared by analyzing the current situation of the dissolution test methods adopted in consistency evaluation for generics and the development and validation of dissolution test methods in USP were focused on so as to provide suitable dissolution test methods for the consistency evaluation of generics. The dissolution test methods established by referring to the related requirements of USP can effectively improve the compliance and efficiency of the consistency evaluation of generics.
KEY WORDS dissolution; United States Pharmacopoeia; Chinese Pharmacopoeia; consistency evaluation
溶出度試驗是檢驗藥品質量的關鍵項目[1],在仿制藥一致性評價中起著關鍵作用[2],各主要國家的藥典均收載了溶出度試驗的操作方法、裝置、條件和評價等內容,用于規范溶出度試驗的測定和相關質量標準的開發。藥典中有關溶出度試驗的技術要求對開展仿制藥一致性評價有重要的指導意義和參考價值。
國家食品藥品監督管理總局(以下簡稱為“總局”)自2015年以來先后發布了《普通口服固體制劑溶出度試驗技術指導原則》[3]、《藥物溶出度儀機械驗證指導原則》[4]和《普通口服固體制劑溶出曲線測定與比較指導原則》[5]等溶出度相關的技術指導原則,用于指導和規范企業的仿制藥開發及其質量控制。然而調研發現,美國除有類似的技術指導原則指導溶出度試驗外,美國藥典中對溶出度試驗有更為全面的技術規范和指導內容。因此,本文通過分析仿制藥一致性評價中采用的溶出度試驗方法的現狀,比較美國藥典和中國藥典中的溶出度試驗方法,重點介紹美國藥典中有關溶出度試驗方法開發與驗證的內容,以期為仿制藥一致性評價提供溶出度試驗技術指導,也為修訂和完善中國藥典中有關溶出度試驗相關標準提供借鑒和參考。
1 仿制藥一致性評價中采用的溶出度試驗方法的現狀分析
1.1 溶出度試驗方法應用的廣泛性
根據總局2016年5月發布的《總局關于落實〈國務院辦公廳關于開展仿制藥質量和療效一致性評價的意見〉有關事項的公告(2016年第106號)》[6]的要求,2018年底前須完成仿制藥一致性評價的品種共有289種(以下簡稱為“289品種”),其中90%以上為口服固體片劑和膠囊劑。
在289品種中,有223種品種采用溶出度試驗方法進行質量控制,占總品種數的77%,所用溶出度試驗裝置包括籃法、槳法和小杯法,以用槳法的品種數最多,占223種品種的近48%。
1.2 溶出度試驗方法應用的客觀性
289品種均為《國家基本藥物目錄(2012年版)》中的化學仿制藥口服固體制劑。根據國家基本藥物遴選條件,289品種應符合防治必需、安全有效、價格合理、使用方便、中西藥并重、基本保障、臨床首選和基層能夠配備的要求[7],因此這些品種應有可靠的質量保障,制定嚴謹的質量標準是提高它們質量水平的客觀需要。
通過查閱289品種的公開的質量標準發現,多數品種已被2015年版中國藥典收載,僅有30種品種未被中國藥典收載,約占總品種數的10%。依據總局2017年8月發布的《關于企業開展289目錄內仿制藥質量和療效一致性評價基本情況信息》[8],在未被2015年版中國藥典收載的30種品種中,企業暫時全部選擇放棄開展一致性評價的品種為雙氯芬酸鈉緩釋膠囊劑(Ⅲ)和鹽酸克林霉素片,企業暫時選擇不放棄開展一致性評價、但尚未開展評價的品種為環孢素膠囊劑和雙氯芬酸鈉緩釋片(Ⅴ),其余26種品種均有企業在開展一致性評價。其中,地紅霉素腸溶片和地紅霉素腸溶膠囊劑尚無公開的質量標準可作參考;聯苯雙酯片和制霉素片的公開的國家標準分別在化藥地標升國標第十冊和部頒抗生素藥分冊中,但標準中無溶出度或崩解時限檢查項,而持有聯苯雙酯片批準文號的企業數有61家,開展一致性評價的企業數為12家,持有制霉素片批準文號的企業數有18家,開展一致性評價的企業數為6家。開展一致性評價的企業數較多,制定適宜的溶出度質量標準能為企業提供參考和指導,是開展一致性評價的客觀需要。endprint
1.3 溶出度試驗方法應用的必要性
依據總局發布的《化學藥品仿制藥口服固體制劑質量和療效一致性評價申報資料要求(試行)》[9],對制劑質量控制的分析方法,須提供質量標準中各項目的檢查方法及其篩選、優化過程并與其他主要藥典收載的試驗方法進行列表比較。本文統計了289品種中被2015年版中國藥典收載、但無溶出度檢查項的品種,并列出了美國藥典、英國藥典和日本藥典中同品種的溶出度試驗方法和規定限度(表1)。其中熊去氧膽酸片,美國藥典、英國藥典和日本藥典收載的該品種的質量標準中均有溶出度檢查項,但中國藥典中無此檢查項。經查閱發現,熊去氧膽酸為生物藥劑學分類系統分類2類藥物[10],溶出可能為體內吸收限速步驟,在其質量標準中設置溶出度檢查項更有利于該品種的質量控制,而參考國外藥典標準可為該品種溶出度試驗方法的建立與開發提供參考和依據。
依據總局仿制藥質量與療效一致性評價辦公室發布的《關于公開征求〈可豁免或簡化人體生物等效性試驗(BE)品種〉意見的通知》[11],擬對289品種中的58種品種予以豁免或簡化人體生物等效性試驗,對其中20種品種擬采用藥學方法評價一致性,豁免生物等效性試驗;另20種品種也擬采用藥學方法評價一致性,但企業須自證屬生物藥劑學分類系統分類1或3類藥物,并依據《人體生物等效性試驗豁免指導原則》提交溶解度、滲透性和溶出度等相關研究資料。由此可見,對擬申請生物等效性試驗豁免的品種,溶出度是仿制藥一致性評價的必要質量控制手段。
2 中美藥典中有關溶出度試驗的內容及比較
美國藥典中除有通則711溶出度和通則724藥物釋放度外,還有通則1 092溶出度試驗開發與驗證、通則1 094膠囊劑溶出度測定與相關質量屬性和通則1 087固有溶出度——轉碟法與固定碟法的溶出度試驗方法3個通則。通則1 094討論了影響膠囊劑溶出度測定結果的相關質量性質,包括膠囊劑類型、生產和包裝對溶出度測定的影響以及明膠膠囊的交聯反應、溶出度測定方法設計、方法學驗證、開始點的建議和關鍵質量性質6部分內容,為膠囊劑溶出度試驗方法的開發與驗證提供了參考。通則1 087討論了表面積固定的非崩解壓縮物在給定溶劑介質中溶出速率的測定方法,固有溶出速率測定在新化學實體藥品開發過程中有時可以用來預測生物利用度,因此非常重要。通則1 092介紹了溶出度試驗方法的開發與驗證過程,對口服固體制劑開發具有廣泛的指導意義和實用價值,稍后將作重點介紹。
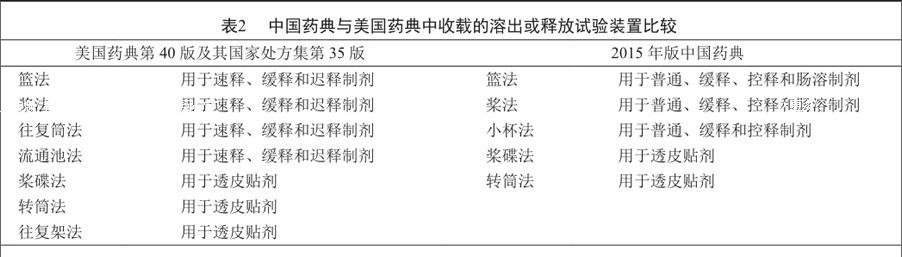
美國藥典中的溶出度試驗方法包括通則711和724,通則711與歐洲藥典、日本藥典的相應內容基本一致,介紹了通用性溶出度試驗裝置、測定方法和結果判定等內容;通則724主要介紹了用于測定透皮貼劑的藥物釋放或溶出的試驗裝置、測定方法和結果判定等內容。美國藥典中收載的用于溶出或釋放試驗的裝置有7種,中國藥典收載了5種,未收載的是往復筒法和流通池法(表2)。從結果判定來看,中國藥典與美國藥典存在一定的差異。中美藥典對溶出規定限度(quantity, Q值)的定義不同,中國藥典中的Q值通常指溶出規定限度,而美國藥典中的Q值為進一步確定溶出規定限度的數值,并不是指溶出規定限度本身。
2015年版中國藥典將溶出度和釋放度的測定方法合并為一個通則(通則931溶出度與釋放度測定法)。隨著總局加入人用藥品注冊技術要求國際協調會議,中國藥典與美國藥典、歐洲藥典和日本藥典等國外主要藥典中的溶出度相關內容的協調適應有了外部條件的支持,區分溶出試驗的通用性方法和特殊性方法更有利于與國際接軌。
3 美國藥典中的通則1 092的主要內容介紹
美國藥典中的通則1 092介紹了溶出度試驗方法的開發與驗證過程,主要用于口服固體制劑。該通則的主要內容包括:①前期評估;②方法的開發;③分析階段;④自動化;⑤驗證;⑥可接受標準。
3.1 前期評估
前期評估主要從原料藥、濾膜、介質和裝置4個方面進行考慮。
測定原料藥的理化特性是選擇合適的溶出介質的步驟之一。要確定溶出介質的組成,重要的是先評估緩沖液及其pH以及不同的表面活性劑(如需要的話)對藥品的溶解度和穩定性的影響。在確定藥品活性成分穩定性滿足測定方法要求的前提下,推薦選擇不少于3種不同pH如pH 1.2、4.5和6.8的溶出介質進行溶出曲線考察。
過濾是樣品處理的關鍵步驟,目的是去除溶液中未溶解的藥物和輔料。過濾材料必須與溶出介質和藥物相容,孔徑范圍一般為0.20 ~ 70 μm。需評估過濾器對藥物的吸附情況。由于過濾材料可能吸附藥物,因此應對過濾材料影響藥物回收率的情況進行具體分析。濾出液中因過濾器而損失的藥物百分比可能取決于藥物濃度,因此應評估樣品溶液在預期濃度范圍內不同濃度下的吸附干擾程度。
開發的溶出度試驗方法應滿足漏槽條件,即溶出介質體積至少為藥物飽和溶液體積的3倍。常用的溶出介質有稀鹽酸溶液、pH 1.2 ~ 7.5的磷酸鹽或醋酸鹽緩沖液、模擬胃或腸液和水。籃法和槳法的溶出介質體積一般為500 ~ 1 000 ml。
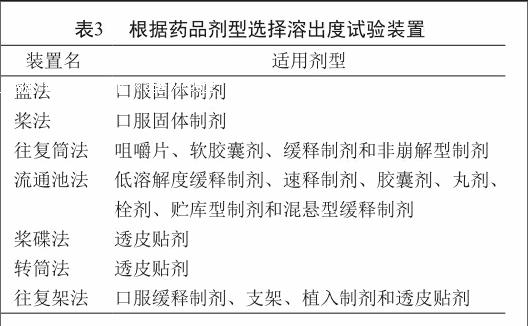
溶出度試驗裝置的選擇須考慮藥品的處方和制劑的體外行為,通常首選藥典通則推薦的裝置(表3)。
3.2 方法的開發
在開發溶出度試驗方法過程中,應研究變異產生的來源并盡可能降低變異性,開發內容則主要包括脫氣、沉降籃、攪拌轉速、研究設計、數據處理和溶出方法評估。endprint
氣泡可能會阻礙溶出,從而影響試驗結果的可靠性。難溶性藥物對氣泡的干擾最為敏感,因此測定此類制劑的溶出度時須考慮脫氣,典型的脫氣方法包括加熱、過濾和短時間抽真空。
采用籃法測定溶出度時,沉降籃常用于調整可能會漂浮的制劑的浮力。通常,沉降籃用于維持制劑在溶出杯的底部,也用于避免制劑(如薄膜衣片)黏附于溶出杯壁。
對速釋膠囊劑或片劑,采用籃法時的攪拌轉速常為50 ~ 100 r/min,采用槳法時的攪拌轉速常為50或75 r/min,使用其他攪拌轉速應提供相應的依據。采用往復筒法時使用的浸率范圍為5 ~ 30浸/min。采用流通池法時的標準流速為4、8或16 ml/min。
研究設計包括取樣時間點、觀察、取樣和清洗4部分內容。①溶出時間點多通過對溶出曲線數據的評估來確定。速釋制劑的溶出時間通常為30 ~ 60 min,多數情況下采用單點測定即可滿足藥典的要求。對緩釋制劑,至少應取3個時間點測定以防劑量傾瀉,需達到完全釋放來繪制體外釋放曲線(>80%);對遲釋制劑,通常需進行至少2個時間點的測定,因此在開發階段應評估整個溶出曲線數據。相似因子不適用于15 min內溶出超過85%的制劑。采用相似因子法比較溶出曲線相似性時需有多個時間點,當平均溶出度(n=12)低于85%時至少應取2個時間點,而在平均溶出度超過85%時可只取一個時間點。②目測觀察和記錄制劑的溶出或崩解行為能夠預測藥品處方或生產過程中的變量,在溶出度試驗方法開發和藥品處方優化過程中特別有用。③取樣位置必須符合藥典的要求。不優選補液,因為制劑單位可能會在溶出介質轉運過程中受到干擾,但當不滿足漏槽條件時則必須進行補液。④應重點評估試驗之間的清洗過程。改變溶出介質和(或)制劑時必須進行清洗,殘留在溶出杯中的物質會影響試驗結果。
溶出度試驗方法由試驗裝置、溶出介質和試驗條件組成,其應對關鍵質量屬性的變化敏感,且耐用性強、重復性好,可用于日常測定并在各實驗室使用。
溶出度試驗結果通過累積速率或分數速率來評估。國內一般采用累積速率來評估,數據處理時需考慮一些相關因素(表4)。
3.3 分析階段
分析階段包括樣品處理和分析過程。分光光度法和高效液相色譜(high performance liquid chromatography, HPLC)法是最常用的分析方法。分析階段的主要內容包括樣品處理、過濾、離心、分析方法、分光光度法或HPLC法測定。
從溶出介質抽取出的樣品需進一步處理以滿足分析方法測定的要求,如過濾除去未溶解的樣品顆粒或避光、低溫貯存樣品等。
過濾應重點考察濾膜的相容性。
不優選對樣品進行離心操作,原因是在藥物固體移除之前,藥物微粒與上清液會形成濃度梯度,由此產生的能量可能導致藥物微粒進一步溶出。
溶出樣品的含量測定常用分光光度法或HPLC法,兩法各有優勢(表5)。
3.4 自動化
自動化的溶出試驗系統可以多種方式和水平進行配置,試驗的準備、取樣和設置取樣點、清洗均可實現自動化,主要內容包括溶出介質的配制、樣品引入和計時、取樣和過濾、清洗、操作軟件和計算結果、常見偏差分析。
自動化的溶出試驗系統所用的溶出介質一般通過稀釋濃縮物來制備。濃縮物的理化穩定性以及使用期間稀釋液的均一性是重要的考慮因素。
自動化的樣品導入和試樣移取較手動更有優勢,能減少試驗間隔期溶出杯之間記時的變異性。
自動取樣可替代手動取樣,尤其適合于多個時間點的取樣。籃法和槳法的取樣位置應在攪拌裝置的頂端至溶出介質表面的中點。取樣探針應從取樣位置取樣。多個取樣時間點的消耗取樣可用等體積的新溶出介質進行補充。當取樣量體積超過溶出介質總體積的1%時對測定的影響較大。取樣和移液設備對溶出藥物的吸附作用也需考慮。
推薦在取樣時間之間和管路運行狀態時評估清洗和沖洗的有效性。
3.5 驗證
試驗方法驗證的主要內容包括專屬性、線性及其范圍、精密度、準確度或回收率、耐用性、樣品及標準溶液的穩定性,參考了美國藥典中和人用藥品注冊技術要求國際協調會議的有關分析方法驗證的內容,此處不作詳述。
3.6 可接受標準
可接受標準和時間點應有區分力。溶出度試驗結果可用于進行關鍵臨床試驗申請的數據之一,當溶出速率顯著影響生物利用度時,溶出度試驗結果和可接受標準應能區分不可接受的生物利用度的藥品批次。當藥品處方和生產工藝的變化顯著影響藥物溶出且這些變化不受質量標準控制時,溶出度試驗結果和可接受標準應能區分這些變化。
溶出度試驗結果的可接受標準是Q值,表示藥品在特定時間內溶出的標示百分比。速釋制劑的Q值一般為75% ~ 80%,通常不超過80%。
遲釋片劑或膠囊劑的溶出度試驗分為兩部分,第一部分制劑暴露在酸性介質中,第二部分制劑暴露在緩沖液介質中。酸性介質常用0.1 mol/L的鹽酸溶液,一般在酸性介質中放置2 h;緩沖液介質常用pH 6.8的磷酸鹽緩沖液,一般在緩沖液介質中放置45 min。放置的時間應依據制劑特性而定。對遲釋制劑,應通過評估整個溶出曲線來確定Q值和取樣時間點。
緩釋制劑溶出度試驗的持續時間更長,至少應選取3個特定的時間點。早期時間點為1 ~ 2 h,指示是否有劑量傾瀉的可能;中間時間點定義制劑的體外釋放曲線;最終時間點用來指示藥物是否完全釋放。應通過評估整個試驗持續時間的溶出曲線來確定時間點。endprint
4 結語
美國藥典中的通則1 092對藥品溶出度試驗方法的開發與驗證有廣泛的指導意義。隨著國內仿制藥一致性評價推進的深入,采用國際通行的標準和規范構建完善的溶出度相關標準及試驗方法有助于促進仿制藥的開發、提高仿制藥的質量、提高仿制藥一致性評價的通過概率,也有助于企業在國內外同步申報。
參考文獻
[1] Hanson R, Gray V. 溶出度試驗技術[M]. 寧保明, 張啟明,譯. 3版. 北京: 中國醫藥科技出版社, 2007: 1-201.
[2] Anand O, Yu LX, Conner DP, et al. Dissolution testing for generic drugs: an FDA perspective [J]. AAPS J, 2011, 13(3): 328-335.
[3] 食品藥品監管總局. 國家食品藥品監督管理總局關于發布普通口服固體制劑溶出度試驗技術指導原則和化學藥物(原料藥和制劑)穩定性研究技術指導原則的通告(2015年第3號)[EB/OL]. (2015-02-05) [2017-11-17]. http://www. sda.gov.cn/WS01/CL0087/114286.html.
[4] 食品藥品監管總局. 總局關于發布藥物溶出度儀機械驗證指導原則的通告(2016年第78號)[EB/OL].(2016-04-28) [2017-11-17]. http://www.sda.gov.cn/WS01/CL0087/151716.html.
[5] 食品藥品監管總局. 總局關于發布普通口服固體制劑參比制劑選擇和確定等3個技術指導原則的通告(2016年第61號)[EB/OL]. (2016-03-18) [2017-11-17]. http://www. sda.gov.cn/WS01/CL0087/147583.html.
[6] 食品藥品監管總局. 總局關于落實《國務院辦公廳關于開展仿制藥質量和療效一致性評價的意見》有關事項的公告(2016年第106號)[EB/OL]. (2016-05-25) [2017-11-17]. http://www.sda.gov.cn/WS01/CL0087/154042.html.
[7] 國家衛生計生委, 國家發展改革委, 工業和信息化部, 等.關于印發國家基本藥物目錄管理辦法的通知[EB/OL].(2015-02-13) [2017-11-17]. http://www.nhfpc.gov.cn/yaozs/ s3581/201504/8147002103b741179217eced1ad77efc.shtml.
[8] 食品藥品監管總局. 關于企業開展289目錄內仿制藥質量和療效一致性評價基本情況信息[EB/OL]. (2017-08-21)[2017-11-17]. http://www.sda.gov.cn/WS01/CL1757/176275. html.
[9] 食品藥品監管總局. 總局關于發布化學藥品仿制藥口服固體制劑質量和療效一致性評價申報資料要求(試行)的通告(2016年第120號)[EB/OL]. (2016-08-16) [2017-11-17]. http://www.sda.gov.cn/WS01/CL1757/163240.html.
[10] Intra-agency agreement between the Eunice Kennedy Shriver National Institute of Child Health and Human Development(NICHD) and the U.S. Food and Drug Administration (FDA). Oral formulations platform — report 1 [EB/OL]. (2016-12-04)[2017-11-17]. https://bpca.nichd.nih.gov/collaborativeefforts/ initiatives/Documents/Formulations_Platform_Report1.pdf.
[11] 仿制藥質量與療效一致性評價辦公室. 關于公開征求《可豁免或簡化人體生物等效性試驗(BE)品種》意見的通知[EB/OL]. (2017-11-11) [2017-11-17]. http://www.cde.org. cn/news.do?method=viewInfoCommon&id=314108.endprint
