溶劑熱法合成Fe-CeO2與N-Fe-CeO2納米粉體及其光催化性能
2018-05-05 06:22:34黃建平折小梅石惠民
無機化學學報 2018年5期
黃建平 陳 芳 折小梅 王 赫 石惠民
(1湖南大學,材料科學與工程學院,長沙 410082)
(2湖南大學,物理與電子科學學院,長沙 410082)
0 引 言
近年來,隨著工業(yè)技術(shù)的發(fā)展,人民生活水平逐步提高,但環(huán)境污染問題也日益嚴峻。光催化技術(shù)是利用光能治理有機污染物、無機污染物和制取氫等的一門新興技術(shù),引起了學者的廣泛關(guān)注。目前,除了技術(shù)相對成熟并且已獲廣泛應用的TiO2外,新型光催化劑也不斷被開發(fā)出來。其中,CeO2由于具有良好的催化活性和穩(wěn)定性,較高的氧化還原性能(Ce3+/Ce4+),且成本低、無毒等特點,在光催化領(lǐng)域的應用前景已倍受關(guān)注[1-3]。與TiO2的寬禁帶,太陽能利用率低,量子效率低等缺點類似[4],CeO2的禁帶寬度約3.2 eV[5],通常只吸收400 nm以下的紫外光,而不能充分利用太陽光中能量集中的可見光。因此唯有降低CeO2的帶隙寬度,使其光響應范圍延伸至可見光區(qū)才能從根本上提高CeO2光催化活性。摻雜是提高CeO2光催化活性的有效途徑之一,通過適當?shù)膿诫s,在CeO2帶隙中引入雜質(zhì)能級以減小其帶隙寬度。目前通過摻雜改善CeO2光催化性能的報道可分為2種:一種是摻雜過渡金屬和稀土元素,如 Fe、Co、Y、La 等[5-12]。Wang 等[7]采用水熱法、共沉淀法和溶劑熱法3種方法合成了Fe摻雜CeO2納米片,發(fā)現(xiàn)在100~175℃的水熱合成的Fe摻雜CeO2對二氯乙烷的光催化性能最好,而175~350℃高溫合成條件下,溶劑熱法制備Fe摻雜CeO2催化性能更佳。袁強等[8]采用溶膠-凝膠法和浸漬法制備了Fe/CeO2固體催化劑,用來催化剛果紅,其最高催化效率達96%。Arul等[9]采用溶劑熱法合成了Fe摻雜CeO2,證實了其對亞甲基藍的降解率得到提高,但并未指出Fe的最佳摻雜量。他們還合成了Co摻雜CeO2,結(jié)果證明Co-CeO2對偶氮染料的光催化性能良好[10]。Liyanage等[5]在CeO2中摻雜不同含量的Y,發(fā)現(xiàn)隨著Y含量的增加,對靛藍胭脂紅、羅丹明B的光催化降解性能都逐步提高。另一種是摻雜非金屬元素,如N、C等[13-19]。顧明杰等[14]以三乙醇胺為氮源,通過溶劑熱法成功制備氮摻雜納米CeO2,并且發(fā)現(xiàn)氮摻雜CeO2對酸性橙的降解率高達93.9%。Mao等[15]以乙二胺為氮源,通過蒸餾回流法成功將氮摻入CeO2,通過可見光催化分解亞甲基藍溶液,發(fā)現(xiàn)CeO2摻雜4%氮時光催化活性最好。Wu等[16]以濃硝酸為氮源,采用溶劑熱法將氮摻入CeO2,結(jié)果顯示摻氮CeO2對羅丹明B的降解率是純CeO2的12.6倍。翦立新等[19]利用微波等離子技術(shù)結(jié)合溶膠凝膠法制備了Fe3+-N-CeO2,發(fā)現(xiàn)當摻Fe量為0.1%時,F(xiàn)e3+-N-TiO2具有最好的催化活性,而N摻雜進一步增強了對可見光的利用率。因此,通過適當?shù)姆绞剑瑩诫s適量的過渡金屬、稀土金屬或非金屬元素,在一定程度上可提高CeO2的光催化性能。但由于金屬元素與非金屬元素在CeO2晶格中摻雜位置不相同,對光催化性能的提高機制也不相同。
因此,為了考察金屬與非金屬元素共同摻雜對CeO2光催化性能的協(xié)同效果,本文以Ce(NO3)3·6H2O為鈰源,F(xiàn)e(NO3)3·9H2O為鐵源,制備了不同含量Fe摻雜CeO2(Fe-CeO2),并用不同含氮溶劑為氮源,制備了不同氮源的N-10%Fe-CeO2納米粉體,并考察了其在模擬太陽光照條件下對有機污染物亞甲基藍(MB)的光催化降解性能。
1 實驗部分
1.1 主要試劑
所用的化學試劑主要有六水合硝酸鈰 (威海佰德信新材料有限公司,分析純,>98%),九水合硝酸鐵(上海阿拉丁生化科技股份有限公司,分析純,>98%),乙二醇(國藥集團化學有限公司,分析純,>99%),濃氨水(成都市科龍化工試劑廠,分析純,24%~26%),尿素(天津市恒興化學試劑制造有限公司,分析純,>99%),三乙醇胺(上海阿拉丁生化科技股份有限公司,分析純,>98%),二乙醇胺(上海阿拉丁生化科技股份有限公司,分析純,>99%),亞甲基藍(天津市恒興化學試劑制造有限公司,指示劑,>98%)。氫氧化鈉(天津市進豐化工有限公司,分析純,>98%)。
1.2 樣品的制備
1.2.1 Fe-CeO2催化劑的制備
以Ce(NO3)3·6H2O為鈰源,乙二醇為溶劑,采用溶劑熱法制備了不同摻雜含量的Fe-CeO2納米粉體。首先按照總物質(zhì)的量為0.01 mol,且nFe/(nFe+nCe)=0%,5%,10%,15%分別計算出 Ce(NO3)3·6H2O 和Fe(NO3)3·9H2O 的質(zhì)量。 先將 Ce(NO3)3·6H2O 溶解于35 mL乙二醇攪拌至溶解后,再加入對應的Fe(NO3)3·9H2O,再攪拌30 min至充分溶解,再加入5%NaOH溶液,調(diào)節(jié)其pH值為9~10,繼續(xù)攪拌30 min。將溶液轉(zhuǎn)移至50 mL聚四氟乙烯內(nèi)襯反應釜中,在180℃的馬弗爐中反應24 h。然后隨爐冷卻至室溫,取出生成的沉淀,用去離子水和乙醇溶液反復洗滌數(shù)遍,離心分離后放入70℃干燥箱中干燥。最后將干燥好的粉體500℃煅燒2 h,即得到不同物質(zhì)的量比的Fe-CeO2納米粉體。
1.2.2 N-Fe-CeO2催化劑的制備
以 Ce(NO3)3·6H2O 為鈰源,F(xiàn)e(NO3)3·9H2O 為鐵源,三乙醇胺(TEA),氨水,尿素(urea),二乙醇胺(DEA)為氮源,按上述nFe/(nFe+nCe)=10%制備出N-10%Fe-CeO2粉體。首先稱取4份3.9 g的Ce(NO3)3·6H2O溶于30 mL乙二醇中,再分別加入0.404 g的Fe(NO3)3·9H2O,攪拌30 min至完全溶解混合,分別加入10.7 mL的三乙醇胺,12 mL濃氨水,4.8 g尿素,7.7 mL二乙醇胺,繼續(xù)攪拌30 min使其充分混合,將溶液轉(zhuǎn)移至50 mL聚四氟乙烯內(nèi)襯反應釜中,在180℃的馬弗爐中反應24 h。樣品的洗滌、干燥與煅燒過程與Fe-CeO2粉體一致。樣品分別標記為 TEA-N-10%Fe-CeO2,NH3·H2O-N-10%Fe-CeO2,Urea-N-10%Fe-CeO2,DEA-N-10%Fe-CeO2。
1.3 樣品的表征
采用D/Max2500型X射線衍射儀(XRD)分析了樣品的物相成分,管電壓為40 kV,電流為10~450 mA, 靶材為銅靶(λ=0.154 nm), 掃描范圍為20°~80°;采用JEM-2100型透射電子顯微鏡(TEM)觀察了樣品的微觀形貌,加速電壓200 kV;采用ORIUS SC1000能量散射X射線光譜分析儀(EDS)分析了樣品的元素分布狀態(tài)。采用ESCALAB 250Xi型X射線光電子能譜(XPS)對樣品的表面化學狀態(tài)進行了分析,X射線光源為200 W單色Al Kα射線;采用LabRAM-010激光拉曼光譜儀對樣品進行拉曼分析,激發(fā)波長為532 nm;采用Agilent Cary 300紫外-可見分光光度計(UV-Vis)測試固體樣品的吸光度。
1.4 粉體的光催化性能及循環(huán)使用性能測試
取50 mg光催化劑分散于50 mL濃度為10 mg·L-1的亞甲基藍(MB)溶液,先在攪拌作用下暗反應30 min,以達到吸附脫附平衡。然后將溶液置于氙燈(PLS-Microsolar 300 W)下照射,持續(xù)不斷電磁攪拌,光源距溶液表面10 cm,光功率密度約為2 000 mW·cm-2。每隔30 min取樣,離心分離得上清液,用TU-1901紫外可見分光儀測試溶液在最大吸收波長664 nm的吸光度。由于吸光度與溶液濃度成正比,遵循朗伯-比爾定律,因此可用吸光度A/A0來表示溶液的濃度變化C/C0,從而評價光催化劑的催化效應。A,C分別為反應時間為t時的吸光度值和溶液濃度,A0,C0分別為暗反應前的吸光度值和溶液濃度。
將每次光催化試驗后的催化劑,離心分離進行回收,使用去離子水反復洗滌多次,烘干后再次測試其光催化性能。如此多次循環(huán)測試,以驗證光催化劑在循環(huán)使用時的催化性能的穩(wěn)定性。
2 結(jié)果與討論
2.1 TEM分析
圖 1(a~d) 分別為純 CeO2、10%Fe-CeO2、NH3·H2O-N-10%Fe-CeO2和Urea-N-10%Fe-CeO2的TEM圖片。從圖中可以看出當添加NaOH為沉淀劑,無氮源時,所得納米顆粒具有較強的團聚性。而濃氨水為氮源時,所獲粉體分散較好,許多粉體堆積形成中空薄壁泡沫狀,而以尿素為氮源時,所獲粉體表現(xiàn)為無序的草葉狀。CeO2基材料的形貌對生長環(huán)境非常敏感,目前已合成了大量形貌各異的CeO2粉體[20]。本實驗中摻雜CeO2形貌主要受pH值變化,氮源的特性及與溶劑的共同作用。根據(jù)其高分辨插圖,測量了其晶面間距為0.306~0.315 nm之間,對應于螢石結(jié)構(gòu)CeO2的(111)面。說明納米顆粒的表面主要是以密排面(111)面為主。對NH3·H2ON-10%Fe-CeO2粉體進行了EDS能譜分析,發(fā)現(xiàn)N、Fe在粉體中的分布比較均勻,也證明了N、Fe在粉體中均勻摻雜。圖1(e~h)分別為圖1(c)中框內(nèi)區(qū)域的Ce、Fe、N和O等元素的分布圖。可以看出這些元素在粉體中均已存在且分布比較均勻。說明Fe、N等元素已均勻摻雜進入了CeO2晶格中。
2.2 XRD分析
利用X射線衍射儀對摻雜Fe、N-Fe的樣品的晶體結(jié)構(gòu)進行了分析,如圖2所示。圖2(a)為不同含量Fe摻雜CeO2的XRD圖。圖中的衍射峰均表現(xiàn)為CeO2的立方螢石結(jié)構(gòu)的特征峰 (PDF No.34-0394)。圖中沒有其它特征峰,表明摻雜的Fe已全部進入CeO2晶格。隨著摻雜Fe含量的增加,衍射峰的位置向高衍射角方向逐漸移動,說明摻雜使得其晶格常數(shù)減小。圖2(b)為10%Fe-CeO2中加入了不同氮源合成的樣品的XRD圖,其衍射峰同樣均表現(xiàn)為CeO2螢石結(jié)構(gòu)的特征峰。衍射峰的位置,半高寬等外形特征基本相似。利用Scherrer公式和(111)、(200)、(220)、(311)的衍射峰的參數(shù)計算了晶粒尺寸,即:


圖 1 (a)純 CeO2、(b)10%Fe-CeO2、(c)NH3·H2O-N-10%Fe-CeO2 和(d)Urea-N-10%Fe-CeO2 的 TEM 圖;(e~h)分別為圖 1(c)中紅框內(nèi)的 Ce、Fe、N、O 的元素分布圖Fig.1 TEM images of(a)pure CeO2,(b)10%Fe-CeO2,(c)NH3·H2O-N-10%Fe-CeO2and(d)Urea-N-10%Fe-CeO2;(e~h)Elements distribution of Ce,Fe,N and O for the area in frame in Fig.1(c)
式中,k為謝樂常數(shù),取值0.89;λ為入射X射線波長,取值為0.154 nm;θ為衍射角;B為實測樣品衍射峰半高寬度。利用布拉格衍射定律和面心立方結(jié)構(gòu)晶面間距的計算公式計算了晶體的晶格常數(shù),如表1所示。晶格常數(shù)隨著摻雜Fe含量的增加不斷減小,主要是由于半徑較小的Fe3+(65 pm)取代了半徑較大的Ce4+(97 pm)的位置。而不同的氮源對其氧化鈰的晶格常數(shù)影響較小,主要是由于N3-(146 pm)離子半徑與O2-(140 pm)半徑接近。晶粒尺寸的變化不大,為10~12 nm之間。說明這種合成方法對晶粒尺寸分布的可控性強。
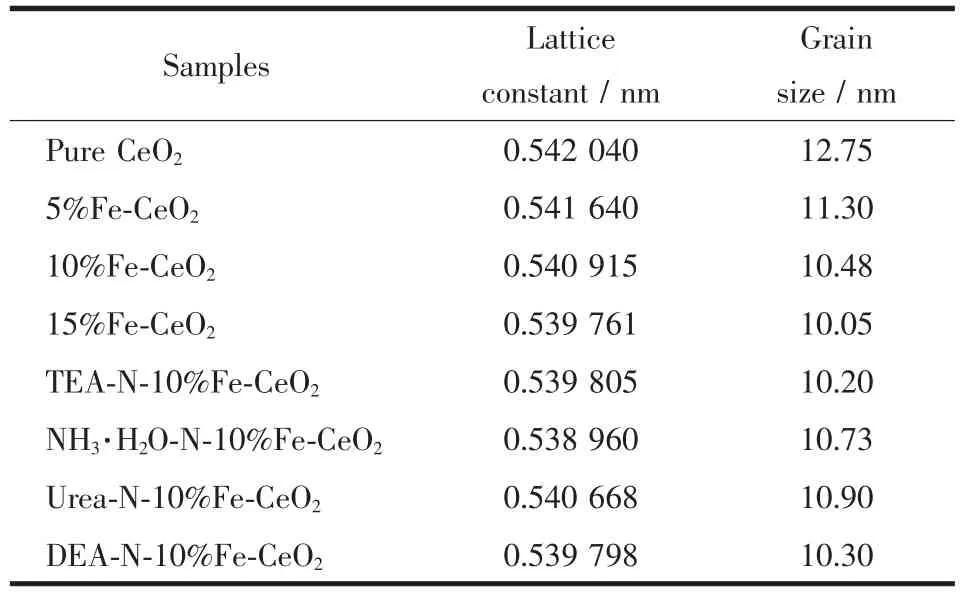
表1 樣品的晶格常數(shù)與晶粒尺寸Table 1 Lattice constants and grain sizes of the samples
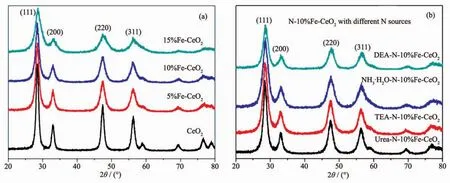
圖 2 Fe-CeO2(a)與 N-10%Fe-CeO2(b)的 XRD 圖Fig.2 XRD patterns of Fe-CeO2(a)and N-10%Fe-CeO2(b)
2.3 XPS分析
圖 3 為 NH3·H2O-N-10%Fe-CeO2與 10%Fe-CeO2中Fe2p、Fe3p和N1s的XPS高分辨圖譜。采用軟件XPSpeak4.1對Fe2p XPS圖譜進行分峰處理,如圖3(a)所示。711.4和717.4 eV分別為Fe2p3/2的主峰及衛(wèi)星峰;724.4和733.2 eV分別為和Fe2p1/2是結(jié)合能主峰及衛(wèi)星峰。衛(wèi)星峰的強度比主峰強度高說明Fe進入CeO2晶格后,F(xiàn)e與Ce之間存在著很強的相互作用。圖3(b)顯示了Fe3p的位置約為 55.6 eV,主要對應為 Fe3+[21]。 圖 3(c)中 399.2和402.9 eV分別代表N在CeO2晶格中和吸附的N2的結(jié)合能峰[18-19]。通過對比,容易看出,濃氨水摻雜后,CeO2晶格中成功摻雜了一定量的N。根據(jù)各元素結(jié)合能峰可估算出10%Fe-CeO2中nFe/(nFe+nCe)=9.2%,NH3·H2O-N-10%Fe-CeO2中 nFe/(nFe+nCe)=5.9%,nN/(nO+nN)=0.9%。
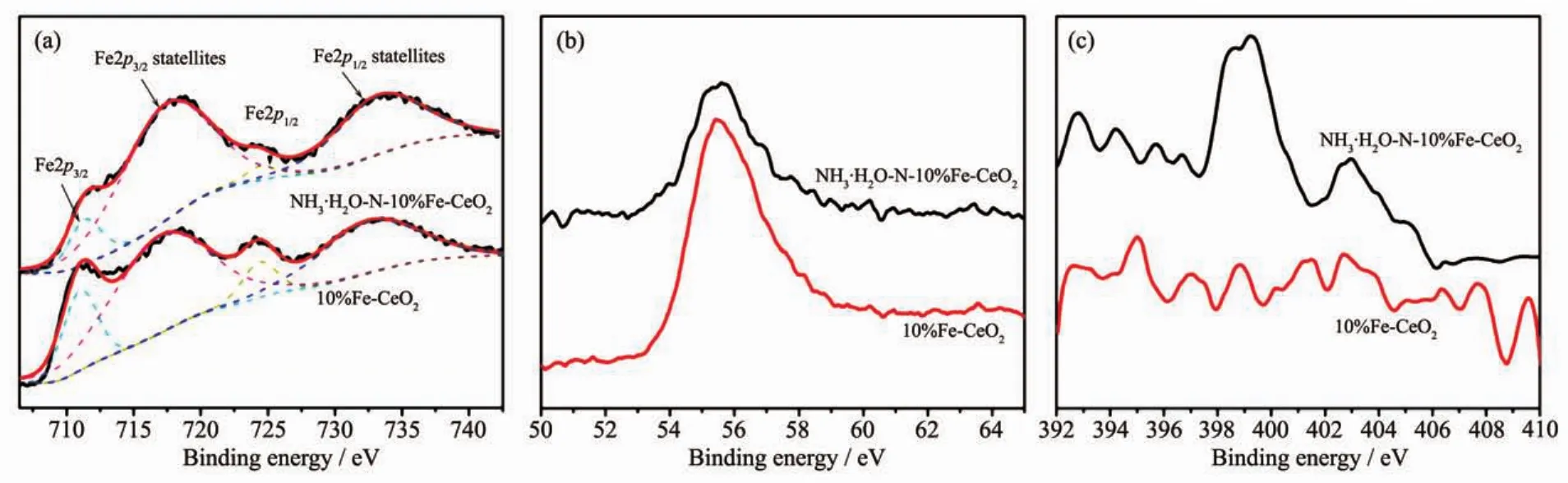
圖 3 NH3·H2O-N-10%Fe-CeO2及 10%Fe-CeO2的 Fe2p(a)、Fe3p(b)和 N1s(c)的 XPS 圖譜Fig.3 Fe2p(a),Fe3p(b)and N1s(c)XPS spectra of NH3·H2O-N-10%Fe-CeO2and 10%Fe-CeO2
2.4 Raman分析
圖4 顯示了不同含量Fe摻雜CeO2與不同氮源的N-10%Fe-CeO2的Raman圖譜。純CeO2的Raman圖譜在462.4 cm-1處存在一個F2g模式振動峰。隨著摻雜Fe含量的增加,F(xiàn)2g峰的位置稍有紅移,強度也逐漸降低,隨著摻雜Fe含量的增加,極化率逐漸降低,以致當Fe摻雜含量達到15%時,其F2g近乎消失。N摻雜也對F2g峰的位置和強度也有明顯的影響,如圖4(b)所示。這說明Fe、N的摻雜改變了CeO2晶格中Ce-O鍵的鍵能和原子間距,導致電子云發(fā)生了遷移。同時,F(xiàn)e、N的摻雜也對晶格的極化率發(fā)生變化。另外,Raman圖譜在550~600 cm-1之間還存在一個比較寬的峰,此峰的形成反應了晶體中的氧空位濃度的變化[22-23]。
2.5 紫外-可見光譜分析
圖5(a,b)分別為不同物質(zhì)的量分數(shù)的Fe摻雜CeO2和不同氮源N-10%Fe-CeO2的紫外-可見光漫反射光譜圖。可見純CeO2的吸收帶邊在400 nm附近垂直陡峭,而摻雜Fe以后,吸收帶發(fā)生明顯紅移,吸收帶邊擴展到可見光波段,緩慢降低。N的摻雜對10%Fe-CeO2有不同程度的影響,以濃氨水做氮源時擴展最大。利用半導體材料帶隙計算公式:

圖4 (a)Fe-CeO2與(b)N-10%Fe-CeO2的Raman圖譜Fig.4 Raman spectra of(a)Fe-CeO2and(b)N-10%Fe-CeO2
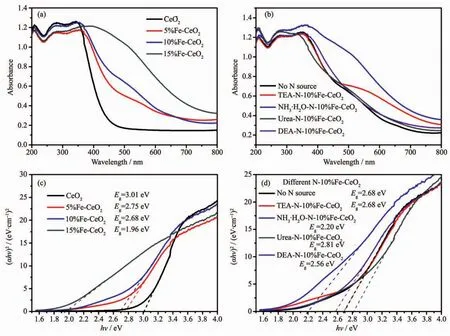
圖5 不同F(xiàn)e含量的Fe-CeO2(a)與不同氮源N-10%Fe-CeO2(b)的UV-Vis吸收圖及(αhν)2~h關(guān)系圖(c,d)Fig.5 UV-visible spectroscopy of Fe-CeO2(a)and N-10%Fe-CeO2(b)and their(αhν)2~h plots(c,d)

式中,α為吸光系數(shù);h為普朗克常數(shù);ν為光的頻率;A為半導體材料的常數(shù);Eg為半導體的禁帶寬度;n為常數(shù),通常材料帶寬為直接型的n取1,間接型的n取4。氧化鈰屬于直接型帶寬材料,因此n取 1。作(αhν)2~hν關(guān)系曲線,如圖 5(c,d)所示。 曲線的切線與橫坐標的截距即為該材料的帶隙寬度Eg。從圖中可見,隨著摻雜Fe含量的增加,帶隙寬度逐漸變小。據(jù)文獻[24]報道,F(xiàn)e摻雜使得導帶寬度增加,導帶底下移,并且在導帶底附近形成了一個局域能級,在原來的禁帶中也產(chǎn)生一個雜質(zhì)能級,從而導致了禁帶寬度變窄。而N的摻雜對10%Fe-CeO2的能帶結(jié)構(gòu)也有一定的微調(diào)作用,主要影響了其對可見光的吸收(圖5(b))。濃氨水做N源時帶隙寬度降低至2.20 eV,而尿素做氮源時,帶隙寬度反而升高至2.81 eV。這說明不同的氮源摻雜對晶體的電子結(jié)構(gòu)起到非常重要的影響。N摻雜進入CeO2晶格中,取代了O的位置。N比O更容易失去電子,因此降低了導帶能級的高度。N的2p電子層與周圍O的2p電子層形成雜化,在價帶頂部產(chǎn)生新的雜質(zhì)電子軌道,縮小了禁帶寬度,從而促進了光生電子與空穴的產(chǎn)生。
2.6 光催化性能分析
圖6為亞甲基藍溶液在氙燈的照射下的光催化降解濃度變化曲線。可見在Fe-CeO2樣品中,以10%Fe-CeO2的樣品催化效率最高,摻雜Fe含量增加到15%后,催化速度反而有所減弱。但最終Fe-CeO2的降解率都在95%左右。而純CeO2在同樣條件下對亞甲藍的降解率只有67%。摻雜N以后,10%Fe-CeO2的催化速率和催化效果產(chǎn)生了不同的變化。以濃氨水和二乙醇胺做氮源時,催化速度進一步提高,最終降解率達到97%。而尿素做氮源時反而使得其催化效果有所降低。這顯然與N摻雜后,其禁帶寬度的改變密切相關(guān)。禁帶寬度越窄,可吸收的紫外線和可見光越多,產(chǎn)生的光生電子-空穴對越多,催化降解亞甲基藍速度越快,最終降解率也越高。由于Fe3+具有未填滿的d層電子軌道,容易捕獲光電子成為Fe2+,也可捕獲空穴成為Fe4+。因此當Fe摻雜含量較低時,可成為光生電子或空穴的陷阱,促進光生載流子的分離,從而起到提高催化性能的作用。然而當Fe摻雜含量過高,F(xiàn)e可能會成為光生電子與空穴的復合中心,雖然禁帶寬度變得更小,但其催化速率反而會降低[18-19]。氮摻雜后,N2p軌道與O2p軌道在導帶底部形成雜化軌道,產(chǎn)生雜質(zhì)能級,降低了導帶能級高度,同時也在價帶頂部形成局部能級[17]。但由于摻雜的氮源不同,N的摻雜含量和摻雜狀態(tài)不同導致活性點數(shù)量和活性點的催化能力不一樣,導致其催化性能也有所不同。摻雜Fe和N對CeO2電子結(jié)構(gòu)的改變和相應的催化機理可用圖7表示。不同的摻雜效應對10%Fe-CeO2的能帶結(jié)構(gòu)產(chǎn)生了不同的影響,從而影響了其最終的催化效果。Fe與N摻雜逐漸降低了導帶的高度,并且在導帶底或價帶頂部形成局域能級,或在帶隙中形成雜質(zhì)能級,從而促進了光生電荷的產(chǎn)生。
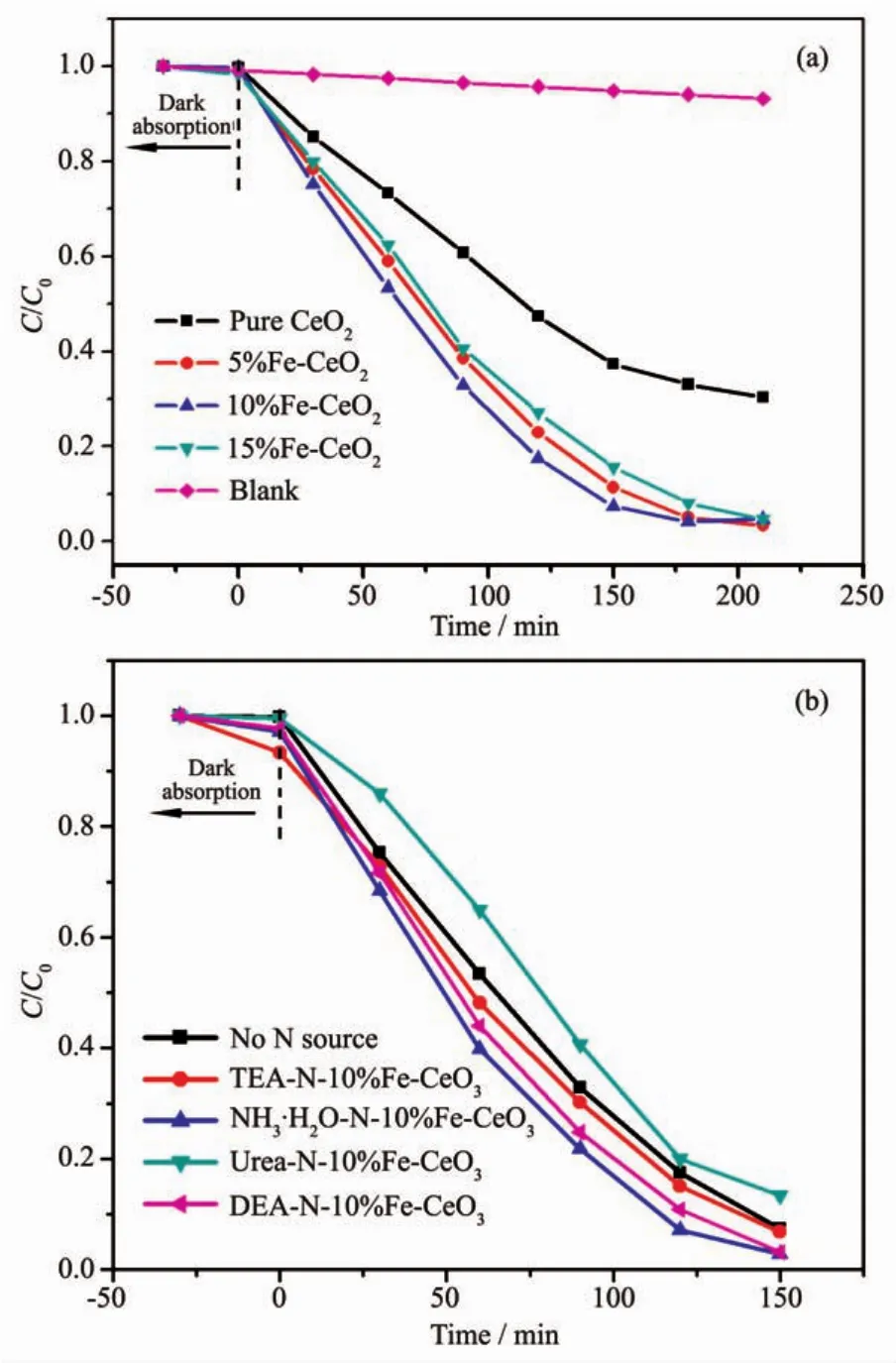
圖6 CeO2和不同含量Fe摻雜的Fe-CeO2(a)及不同氮源N-10%Fe-CeO2(b)作催化劑的光催化降解亞甲基藍溶液性能Fig.6 Photocatalytic degradation of methylene blue solution with pure CeO2and Fe-CeO2as catalystic agents(a)and different nitrogensource N-10%Fe-CeO2as catalytic agents(b)
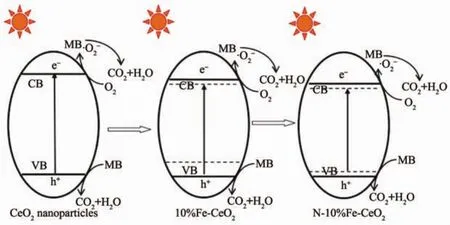
圖 7 CeO2、10%Fe-CeO2和 N-10%Fe-CeO2的催化機理示意圖Fig.7 Schematic diagram of catalytic mechanism of CeO2,10%Fe-CeO2and N-10%Fe-CeO2
2.7 光催化劑的循環(huán)使用性能
圖8 為NH3·H2O-N-10%Fe-CeO2的循環(huán)使用的降解率結(jié)果(每次光催化時間為210 min)。降解率隨著使用次數(shù)的增加,略有降低。經(jīng)過5次使用后,光催化劑在210 min內(nèi)對亞甲基藍的降解率仍有89%,表明催化劑的光催化性能具有良好的穩(wěn)定性。圖8(b)對比了5次循環(huán)使用后的催化劑與制備態(tài)的催化劑的XRD圖,并沒有發(fā)現(xiàn)有明顯變化。說明催化劑在使用前后結(jié)構(gòu)沒有改變,也幾乎沒有吸附其它物質(zhì)。 因此,NH3·H2O-N-10%Fe-CeO2的光催化性能總體上比較穩(wěn)定。在循環(huán)使用過程中,降解率有所下降的原因可能是催化劑在回收再利用過程中發(fā)生了微量的質(zhì)量損失。
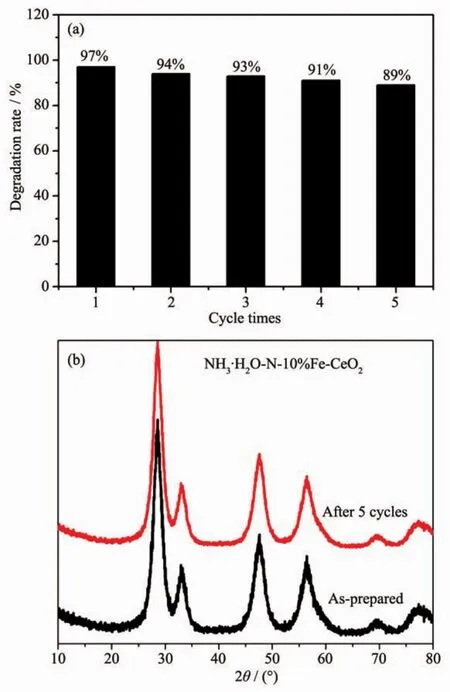
圖8 (a)NH3·H2O-N-10%Fe-CeO2催化劑對亞甲基藍溶液的降解率與循環(huán)使用次數(shù)的關(guān)系;(b)循環(huán)使用前后的XRD對比圖Fig.8 (a)Relationship of photocatalytic degradation rate of methylene blue solution and use time with NH3·H2O-N-10%Fe-CeO2as catalytic agent;(b)XRD patterns comparison for as-prepared and after 5 cycles photocatalysis
3 結(jié) 論
本文采用溶劑熱法合成了不同F(xiàn)e摻雜含量的Fe-CeO2納米粉體及不同氮源摻雜的N-10%Fe-CeO2納米粉體,并對其光催化性能進行了研究。可得到以下結(jié)論:
(1)Fe摻雜引起晶格畸變,形成反應活性中心,產(chǎn)生雜質(zhì)能級,降低禁帶寬度。10%Fe-CeO2的催化速率最高,將純CeO2對亞甲基藍的降解率從67%提高到95%。
(2)摻雜過量Fe時,F(xiàn)e會成為光生電子與空穴的復合中心,從而降低光生載荷的量子效率,降低其催化活性。
(3)不同氮源摻雜對10%Fe-CeO2的生長形貌具有重要影響,其中以濃氨水為氮源摻雜形成中空薄壁狀N-10%Fe-CeO2粉體的禁帶寬度最小,對亞甲藍溶液的降解效率可提高到97%。
(4)以濃氨水為氮源制備的N-10%Fe-CeO2催化劑具有較好的性能穩(wěn)定性。經(jīng)5次循環(huán)使用,對亞甲基藍溶液的光催化降解率仍高達89%。
參考文獻:
[1]SU Lin-Feng(蘇琳峰),GONG Jin-Feng(鞏金峰),MENG Fan-Ming(孟 凡 明).Chinese Journal of Synthetic Chemistry(合成化學研究),2016,4(4):29-37
[2]QI En-Lei(齊恩磊),MAN Li-Ying(滿麗瑩),WANG Sun-Hao(王孫昊),et al.Chinese Journal of Materials Research(材料研究學報),2011,25(2):219-224
[3]ZHAO Xiao-Bing(趙曉兵),YOU Jing(游靜),LU Xiao-Wang(陸曉旺),et al.J.Inorg.Mater.(無機材料學報),2011,26(2):159-164
[4]WU Jun-Ming(吳俊明),WANG Ya-Ping(王亞平),YANG Han-Pei(楊漢培),et al.Chinese J.Inorg.Chem.(無機化學學報),2010,26(2):203-210
[5]Liyange A D,Perera S D,Tan K,et al.ACS Catal.,2014,4(2):577-584
[6]WANG Jing-Xin(王敬欣).J.Chin.Rare Earth Soc.(中國稀土學報),2007,25(S1):82-88
[7]Wang W,Zhu Q,Dai Q G,et al.Chem.Eng.J.,2017,307:1037-1046
[8]YUAN Qiang(袁強),JIANG Ling(江玲),LI Hui(李輝),et al.Journal of Xiamen University:Natural Sciences Edition(廈門大學學報:自然科學版),2011,50(1):70-75
[9]Arul N S,Mangalaraj D,Chen P,et al.Mater.Lett.,2011,65:3320-3322
[10]Arul N S,Mangalaraj D,Han J.Mater.Lett.,2015,145:189-192
[11]Xie S L,Wang Z L,Cheng F L,et al.Nano Energy,2017,34:313-337
[12]ZHANG Guo-Fang(張國芳),ZHANG Yang-Huan(張羊換),GE Qi-Lu(葛啟錄),et al.Spectrosc.Spectr.Anal.(光譜學與光譜分析),2011,31(12):3315-3318
[13]LI Chang-Quan(李長全),LUO Lai-Tao(羅來濤),XIONG Guang-Wei(熊光偉).Acta Chim.Sin.(化學學報),2010,68(10):1023-1026
[14]GU Ming-Jie(顧明杰),LI Rui-Xing(李銳星),SONG Xiao-Zhen(宋曉貞).Journal of Ceramics(陶瓷學報),2013,34(2):135-138
[15]Mao C J,Zhao Y X,Qiu X F,et al.Phys.Chem.Chem.Phys.,2009,40(3):5633-5638
[16]Wu C L.Mater.Lett.,2015,139:382-384
[17]Ren R K,Zhang M J,Meng J,et al.Chin.Phys.B,2017,26(3):036102
[18]HUANG Dong-Sheng(黃東生),CHEN Chao-Feng(陳朝鳳),LI Yu-Hua(李玉花),et al.Chinese J.Inorg.Chem.(無機化學學報),2007,23(4):738-742
[19]JIAN Li-Xin(翦立新),YIN Xiao-Qiu(殷小秋),XIANG Jian-Nan(向建南),et al.Journal of Hunan University:Natural Sciences Edition(湖南大學學報:自然科學版),2006,33(1):79-82
[20]Sun C W,Li H,Chen L Q.Energy Environ.Sci.,2012,5:8475-8505
[21]Toru Y,Peter H.Appl.Surf.Sci.,2008,254:2441-2449
[22]Jeyanthi C E,Siddheswaran R,Kumar P,et al.Ceram.Int.,2014,40(6):8599-8605
[23]Xu B,Zhang Q T,Yuan S S,et al.Catal.Today,2017,281(1):135-143
[24]Tian D,Zeng C H,Fu Y C,et al.Solid State Commun.,2016,231/232:68-79
