基于PPI網絡與機器學習的蛋白質功能預測方法
2018-05-21 00:50:08唐家琪吳璟莉
計算機應用 2018年3期
唐家琪,吳璟莉,2,3
(1.廣西師范大學 計算機科學與信息工程學院,廣西 桂林 541004; 2.廣西師范大學 廣西多源信息挖掘與安全重點實驗室,廣西 桂林 541004;3.廣西區域多源信息集成與智能處理協同創新中心,廣西 桂林 541004)
0 引言
蛋白質是執行生物體內各種重要生物活動的大分子,認識其功能對推動生命科學、農業、醫療等領域的發展意義重大。1961年,Anfinsen等[1]提出蛋白質一級序列決定其三維結構、蛋白質三維結構決定其功能的論斷。相對于蛋白質三維結構,一級序列更容易通過生物實驗測得,故早期的蛋白質功能預測方法大都基于序列相似性原理,利用BLAST(Basic Local Alignment Search Tool)[2]和PSI-BLAST(Position-Specific Iterated BLAST)[3]等工具計算功能未知的蛋白質與功能已知的蛋白質之間的序列相似度,若相似度較高則認為其具有相同的功能。然而,近年來的研究表明,序列相似的蛋白質能夠形成不同的三維結構,故其功能不一定相同,而且序列差異較大的蛋白質也可能具有相同的功能[4];因此,基于序列相似性的蛋白質功能預測方法是不可靠的。
隨著越來越多的蛋白質三維結構數據的產生,FATCAT(Functional And Tractographic Connectivity Analysis Toolbox)[5]和PAST(Polypeptide Angle Suffix Tree)[6]等蛋白質三維結構數據庫相繼建立,研究者提出了基于蛋白質三維結構的功能預測方法[7],這類方法通過計算功能未知的蛋白質與功能已知的蛋白質的三維結構相似度來判斷其是否具有相同功能。相對于蛋白質的氨基酸序列,其三維結構更保守穩定[8],故基于三維結構的方法通常比基于序列的方法更準確,但由于已知三維結構的蛋白質數量較少,其應用范圍較窄。
隨著高通量生物實驗技術與蛋白質相互作用(Protein-Protein Interaction, PPI)預測方法[9-10]的發展,產生了海量的、可用于大規模蛋白質功能注釋的PPI數據,基于蛋白質相互作用網絡(簡稱PPI網絡)的功能預測方法深受關注。根據Oliver[11]提出的關聯效應(Guilt-By-Association, GBA),相互作用的蛋白質具有相同或相似的功能,可以通過分析PPI網絡的拓撲結構,根據網絡中已經注釋功能的蛋白質來推測網絡中未注釋功能的蛋白質的功能。Chi等[12]提出余弦迭代算法(Cosine Iterative Algorithm, CIA),其基于蛋白質之間動態相互作用,迭代更新鄰居蛋白質的注釋術語集,估計它們與未注釋蛋白質之間的功能相似性來完成預測。Xiong等[13]采用譜聚類算法將PPI網絡中的蛋白質劃分為若干功能模塊,根據頂點度、緊密度和介數三種中心性指標標注各模塊中的重要蛋白質,再利用基于 Gibbs抽樣的協同分類算法預測蛋白質功能。Wang等[14]針對注釋術語間的功能關聯性,設計了一種基于多標簽學習的蛋白質功能預測算法。Teng等[15]根據相互作用的蛋白質在PPI網絡中的主被動關系將無向的PPI網絡轉化成有向網絡,并通過在有向PPI網絡中傳播基因本體術語(Diffusing GO Terms in the Directed PPI Network, GoDIN)的方法預測蛋白質的功能。Yu等[16]提出一種在混合圖上隨機游走的蛋白質功能預測方法,該方法不僅綜合考慮了直接和間接相互作用信息,還利用功能相似性權重來減少噪聲相互作用的影響。
基于PPI網絡的功能預測效果依賴于網絡的可靠程度。由于生物實驗技術的制約,大多數PPI數據均存在一定程度的噪聲,從而降低了這類方法的預測精度。本文將蛋白質家族(Family)、結構域(Domain)和重要位點(Important Site)信息作為頂點屬性,整合到PPI網絡中以減輕網絡中數據噪聲的影響,并提出了一種基于層次聚類(Hierarchical Clustering, HC)、主成分分析(Principal Component Analysis, PCA)與多層感知器(Multi-Layer Perceptron, MLP)的蛋白質功能預測方法(HC, PCA and MLP based Method, HPMM)。HPMM將蛋白質功能預測轉化成多標簽二分類問題,首先從PPI網絡、蛋白質家族、結構域和重要位點中提取蛋白質的特征,再訓練MLP模型用于功能預測。采用人類(Homo sapiens)數據集對蛋白質功能預測方法CIA[12]、GoDIN[15]和HPMM進行測試。實驗結果表明,相比CIA和GoDIN,HPMM的精確度與F值更高。
1 問題與符號定義
PPI網絡通常表示為無向圖G(V,E),其中V={v1,v2,…,vn}為頂點集,E={eij|eij=(vi,vj),vi,vj∈V}為邊集。頂點vi(i=1,2,…,n)表示蛋白質,邊eij∈E表示其兩端的蛋白質vi與蛋白質vj之間存在相互作用,di(i=1,2,…,n)表示頂點vi的度,即與蛋白質vi存在相互作用的蛋白質種類數。PPI網絡中,假設v1,v2,…,vn1為功能已知的蛋白質,vn1+1,vn1+2,…,vn1+n2為功能未知的蛋白質,n=n1+n2。用鄰接矩陣An×n表示圖G,其中每個元素aij(i=1,2,…,n,j=1,2,…,n)的取值定義如下:
(1)
蛋白質家族、結構域和重要位點信息對蛋白質的功能有重要影響,故可以看成蛋白質的屬性。用矩陣Pn×m記錄蛋白質屬性,每行表示一個蛋白質頂點,每列表示一個屬性,元素pij(i=1,2,…,n,j=1,2,…,m)的取值定義如下:
(2)
將鄰接矩陣An×n與屬性矩陣Pn×m橫向合并,得到蛋白質的特征矩陣Xn×(n+m),其中xi=(xi1,xi2,…,xi(n+m))為蛋白質樣本vi(i=1,2,…,n)的特征向量,元素xij(i=1,2,…,n,j=1,2,…,n+m)的取值定義如下:
(3)
令Yn×w為記錄蛋白質的功能注釋信息的標簽矩陣,其中w為數據集中功能注釋的類別總數,Yn×w中每一行yi=(yi1,yi2,…,yiw)為蛋白質樣本vi(i=1,2,…,n)的標簽向量,其中的元素yij(i=1,2,…,n,j=1,2,…,w)的取值定義如下:
(4)
根據上述定義,以蛋白質為樣本、功能術語為樣本標簽的蛋白質功能預測問題可轉化為多標簽二分類問題:將n1個功能已知的蛋白質用于訓練預測模型,試圖得到映射函數h:X→Y,使給定功能未知的n2個蛋白質的特征向量xi(i=n1+1,n1+2,…,n),預測其標簽向量(即功能注釋向量)h(xi)?Y。
2 功能預測方法
本章提出一種基于機器學習的蛋白質功能預測方法HPMM,輸入為PPI網絡中功能已知的蛋白質的功能注釋術語,PPI網絡及其每個蛋白質的屬性信息(家族、結構域和重要位點);輸出為PPI網絡中功能未知的蛋白質的功能注釋術語。如圖1所示,HPMM主要分為特征提取、訓練模型和功能預測3個階段。

圖1 HPMM流程 Fig. 1 Flow chart of HPMM
首先基于層次聚類和主成分分析進行特征提取,將提取的功能模塊(Function Module)、屬性(家族、結構域和重要位點)主成分(Principal Component)及頂點度 (Degree) 作為特征,對其歸一化后用于訓練多層感知器,從而得到一個多標簽的二分類模型。然后用該模型預測PPI網絡中功能未知的蛋白質。下面詳細介紹預測方法HPMM的主要步驟。
2.1 功能模塊特征提取
研究表明細胞功能是通過生物大分子之間相互作用形成的功能模塊實現的[17],故同一功能模塊中的蛋白質往往具有相似的功能,因此可先通過聚類算法從PPI網絡中挖掘出若干功能模塊,并將其作為蛋白質的特征以用于功能預測。由于功能模塊特征取決于網絡的整體拓撲結構,其受局部噪聲相互作用的影響較小,魯棒性較強。
層次聚類算法常被用于從PPI網絡中挖掘功能模塊,以確定模塊中蛋白質的功能[18-19]。本文采用Clauset等[20]提出的一種針對復雜網絡的凝聚層次聚類算法提取功能模塊特征。算法輸入為鄰接矩陣An×n,輸出為功能模塊矩陣Fn×k1(k1為功能模塊數),其中的元素fij(i=1,2,…,n,j=1,2,…,k1)取值為1(0),表示蛋白質vi屬于(不屬于)功能模塊j。
算法利用NG(Newman and Girvan)模塊度[21]來評價聚類效果,其定義如式(5)所示:
(5)
其中:fi=(fi1,fi2,…,fik)代表蛋白質vi的功能模塊特征向量,函數δ(fi,fj)指示向量fi和fj取值是否相同:相同返回1,表示蛋白質vi和vj屬于相同功能模塊;反之返回0,表示蛋白質vi和vj屬于不同的功能模塊。模塊度取值越大,則表示圖的模塊劃分效果越好。
2.2 屬性主成分特征提取
如前所述,由于蛋白質的家族、結構域和重要位點這些屬性信息均對其功能起到重要的影響,可將其作為特征以減少預測結果對帶數據噪聲的PPI網絡的依賴,從而提高預測精度;但是,在訓練多層感知器時,若蛋白質樣本維數過多則會使預測模型失去泛化能力,從而影響預測效果。本文考慮到蛋白質屬性之間的相關性,使用一種基于奇異值分解(Singular Value Decomposition, SVD)的主成分分析方法[22],對屬性矩陣Pn×m降維處理。
首先,對Pn×m進行z-score標準化處理,得到矩陣Zn×m,其中每個元素zij(i=1,2,…,n,j=1,2,…,m)取值如下:
(6)
其中:
(7)
然后,對矩陣Zn×m進行奇異值分解,使得Z=UΛΜ,其中U為n階正交矩陣,Λ為n×m的半正定對角矩陣,Μ為m階正交矩陣,U中每列uj(j=1,2,…,n)代表蛋白質屬性的一個主成分,Λ中對角元素λj(j=1,2,…,n)代表對應主成分的方差,該值反映了主成分的重要程度。
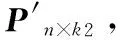
2.3 特征合并與標準化
本文將蛋白質在PPI網絡中的頂點度數作為特征,并將其與功能模塊特征和屬性主成分特征合并,得到特征矩陣Γn×(k1+k2+1),其中元素γij(i=1,2,…,n,j=1,2,…,k1+k2+1)取值如下:
(8)
其中:di表示蛋白質vi的頂點度。該矩陣中包含了功能模塊、屬性主成分、和頂點度三類特征。功能模塊特征反映了蛋白質在宏觀的相互作用網絡中所在的功能模塊。屬性主成分特征則反映了蛋白質微觀層面的信息。頂點度是一種常用的中心性度量,反映了蛋白質在PPI網絡中的重要程度,同時也代表了蛋白質參與生命活動的多少,即功能多樣性[23]。這三類特征從不同層面表征了蛋白質,并且不容易被PPI網絡中數據噪聲干擾。
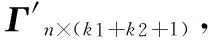
(9)
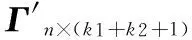
2.4 基于多層感知器的功能預測
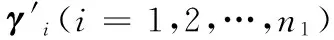
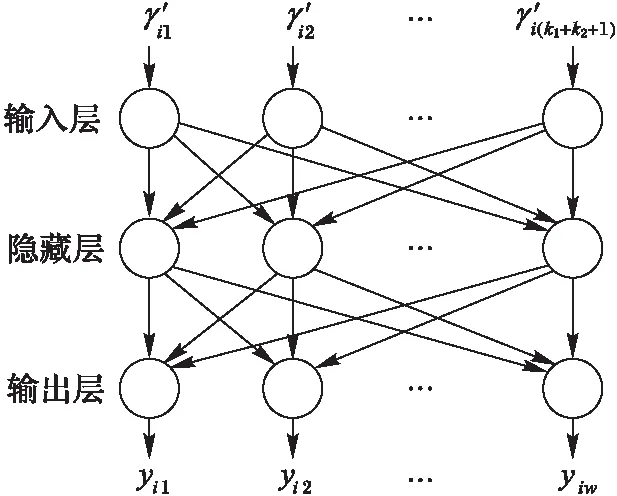
圖2 多層感知器示意圖 Fig. 2 Diagram of MLP
2.4.1 參數設置
輸入層節點數等于特征向量的維數k1+k2+1,輸出層的節點數等于數據集中的所有蛋白質擁有的功能注釋數,即功能注釋向量的維數w。
輸出層使用Sigmoid激活函數,定義如下:
(10)
使用交叉熵(cross entropy)作為輸出層的損失函數,對于樣本vi,其交叉熵定義如下:
(11)


ReLu (x) = max(0,x)
(12)
訓練該神經網絡時,采用批量學習的方式[26],批量大小為訓練集中蛋白質數的10%,迭代次數為400次,學習率(Learning Rate)為0.1,動量(Momentum)為0.9。
2.4.2 功能選擇
3 實驗與結果分析
本文用人類數據集對蛋白質功能預測方法HPMM、CIA[12]和GoDIN[15]進行比較分析。實驗在一臺4核8線程的微型計算機上進行,CPU型號為Intel@Core i7-3630QM 2.4 GHz×8,內存為8 GB,操作系統為Ubuntu 16.04 LTS 64位,編程工具為R 3.3.2。訓練MLP時采用GPU加速技術和MXNet深度學習框架,GPU型號為GeForce GT 650M/PCIe/SSE2。
3.1 實驗數據
本文的實驗數據來自于DIP[27]、基因本體(Gene Ontology, GO)[28]和InterPro[29]數據庫。DIP數據庫提供了人類PPI網絡,GO提供了功能注釋, InterPro數據庫提供了蛋白質家族、結構域和重要位點信息。
首先從DIP數據庫下載人類PPI網絡數據,并用UniProtKB/Swiss-Prot[30]對PPI網絡中的蛋白質進行ID轉換,然后去除網絡中自相互作用、重復相互作用及無法轉換的蛋白質;再通過biomaRt包[31]根據每個蛋白質的UniProtKB/Swiss-Prot編號獲取對應的GO術語編號和InterPro編號。
GO包括分子功能(Molecular Function, MF)、生物過程(Biological Process, BP)和細胞組件(Cellular Component, CC)三個獨立的子本體。為保證注釋術語的可靠性,實驗剔除了獲取手段為IEA(Inferred from Electronic Annotation)、ND(No biological Data Available)和IC(Inferred by Curator)的功能注釋。如上所述,由于MF、BP和CC三個子本體相互獨立,分別為每個子本體建立PPI網絡,分別稱為MF、BP和CC網絡,并刪除了沒有被GO術語注釋的蛋白質。此外,為確保每個蛋白質均有與其相互作用的蛋白質,本文僅取PPI網絡中的極大連通子圖作為測試數據。
InterPro數據庫是一個整合了蛋白質家族、結構域和重要位點信息的綜合數據庫,每個InterPro編號對應一條蛋白質的家族、結構域或重要位點信息。本文根據每個InterPro編號對應的信息存在與否將其編碼成二元變量作為蛋白質頂點的屬性。最終得到三個整合了多元生物信息的PPI網絡,如表1所示。
表1 人類數據集詳情Tab. 1 Details of human data set
3.2 評價指標
本文將精確度(Precision)、召回率(Recall)和F值(F-Measure)作為評價指標來衡量算法的預測效果[32],其定義如式(13)~(15):
(13)
(14)
(15)
其中:TP表示預測的功能術語正確的個數,FP表示預測的功能術語錯誤的個數,FN表示實際的功能術語沒有被預測到的個數。
3.3 性能評價
本節首先給出HPMM在MF、BP和CC三個PPI網絡的功能模塊特征和屬性主成分特征提取情況及MLP節點設置情況,然后對其與CIA[12]和GoDIN[15]的預測效果進行比較分析。
從表2中可以看出,HPMM在三個PPI網絡上提取的功能模塊數和NG模塊度差異不大。
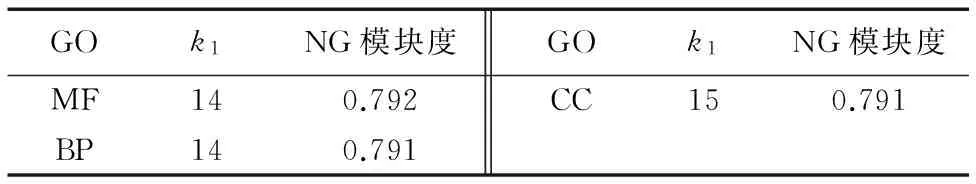
表2 不同PPI網絡的功能模塊特征Tab. 2 Functional module features of different PPI networks
在表3中,三個PPI網絡的蛋白質屬性主成分特征提取結果均接近70%的降維率。例如在MF網絡中,蛋白質屬性特征的個數由806降至243,降維率為69.9%。

表3 不同PPI網絡的屬性主成分特征Tab. 3 Attribute features of different PPI networks
從表4中可以看出,對三個PPI網絡建立的MLP的輸入層的節點數相近。這是因為三個PPI網絡上的蛋白質功能模塊特征數k1和屬性主成分特征數k2接近。此外,由于MLP的輸出層節點數和隱藏層節點數取決于GO術語數,故對BP網絡建立的MLP的輸出層節點數和隱藏層節點數最多,對MF網絡建立的MLP的輸出層節點數和隱藏層節點數介于BP網絡和CC網絡之間,對CC網絡建立的MLP的輸出層節點數和隱藏層節點數最少。

表4 MLP節點設置Tab. 4 MLP nodes setting
采用10折交叉驗證(Ten-fold Cross Validation)來測試HPMM的預測效果。如3.1節所述,MF、BP和CC網絡中的蛋白質均為經過注釋的,為測試功能預測方法的性能,實驗中分別將每個網絡中的蛋白質平均分成10份,輪流將其中9份作為功能已知的蛋白質用于訓練模型,其中1份作為功能未知的蛋白質用于預測,合并10次預測的結果,將預測概率最高的l個GO術語作為蛋白質的功能,對于MF、BP和CC網絡,l分別設為6,15,5。最后將其與真實的功能注釋情況比較。表5給出了HPMM、CIA和GoDIN這3種基于PPI網絡的功能預測方法在MF、BP和CC網絡上的精確度、召回率和F值。從精確度看,HPMM在3個網絡上均高于CIA和GoDIN,并且在MF和CC網絡上優勢明顯。從召回率看,HPMM在3個網絡上均高于CIA,但不如GoDIN。這可能是由于GoDIN方法預測的GO術語較多,以降低精確度為代價提高了召回率。從F值看, HPMM在3個網絡上均高于CIA和GoDIN,并且在MF和CC網絡上優勢明顯。總的來看,HPMM能夠有效預測蛋白質的功能,并在精確度和F值上優于CIA和GoDIN,其優勢在MF和CC網絡上尤為顯著,這可能是由于這可能是由于HPMM考慮的蛋白質屬性(家族、結構域和重要位點)與MF和CC的聯系更緊密,故對其預測效果的提升幅度較大。
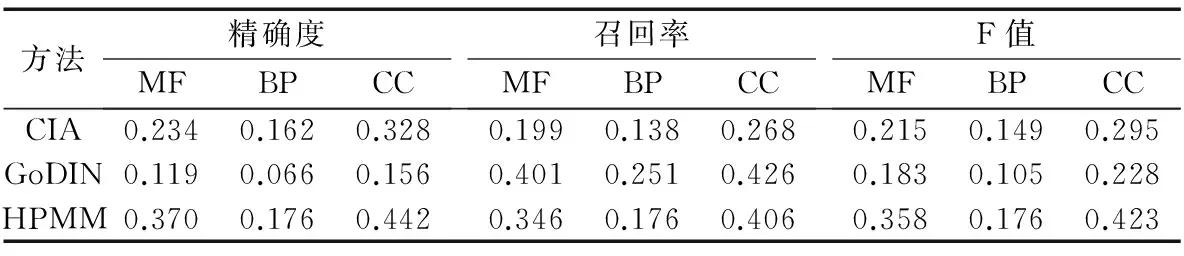
表5 蛋白質功能預測方法性能比較Tab. 5 Performance comparison of protein function prediction methods
4 結語
基于PPI網絡的方法是近年來較為流行的一類蛋白質功能預測方法。這類方法預測成本較低,但其效果容易受PPI網絡中數據噪聲的影響。針對該問題,本文提出了一種基于機器學習的蛋白質功能預測方法HPMM。該方法將蛋白質家族、結構域和重要位點信息整合到PPI網絡中,綜合考慮蛋白質的微觀信息和宏觀相互作用以減輕網絡中數據噪聲的影響,并結合了層次聚類、主成分分析和多層感知器三種機器學習技術來預測蛋白質的功能。為驗證HPMM的有效性,從DIP數據庫下載人類PPI網絡,從InterPro數據庫獲取蛋白質家族、結構域和重要位點對應的編號,并采用了GO功能注釋方案。實驗結果證明該方法能有效預測蛋白質的功能,并且在精確度與F值上優于CIA和GoDIN這兩種完全基于PPI網絡的方法。對于蛋白質功能預測今后的研究, 我們認為可以從以下幾個方面入手:1)通過鏈路預測與圖重構技術建立更具有生物統計特性的PPI網絡以降低數據噪聲的影響。2)深入研究PPI網絡拓撲結構,綜合考慮網絡的局部特性與全局特性用于功能預測。3)研究整合多元生物數據的方法以提升預測效果。
參考文獻(References)
[1] ANFINSEN C B, HABER E, SELA M, et al. The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain [J]. Proceedings of the National Academy of Sciences of the United States of America, 1961, 47(9):1309-1314.
[2] ALTSCHUL S F, GISH W, MILLER W, et al. Basic local alignment search tool [J]. Journal of Molecular Biology, 1990, 215(3): 403-410.
[3] ALTSCHUL S F, MADDEN T L, SCHFFER A A, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs [J]. Nucleic Acids Research, 1997, 25(17): 3389-3402.
[4] GILKS W R, AUDIT B, de ANGELIS D, et al. Percolation of annotation errors through hierarchically structured protein sequence databases [J]. Mathematical Biosciences, 2005, 193(2): 223-234.
[5] YE Y, GODZIK A. FATCAT: a Web server for flexible structure comparison and structure similarity searching [J]. Nucleic Acids Research, 2004, 32(Web Server issue):W582-W585.
[7] LASKOWSKI R A, WATSON J D, THORNTON J M. From protein structure to biochemical function? [J]. Journal of Structural & Functional Genomics, 2003, 4(2/3):167-177.
[8] WATSON J D, LASKOWSKI R A, THORNTON J M. Predicting protein function from sequence and structural data [J]. Current Opinion in Structural Biology, 2005, 15(3): 275-284.
[9] YOU Z H, LEI Y K, ZHU L, et al. Prediction of protein-protein interactions from amino acid sequences with ensemble extreme learning machines and principal component analysis [J]. BMC Bioinformatics, 2013, 14(S8): 1-11.
[10] WEI L, XING P, ZENG J, et al. Improved prediction of protein-protein interactions using novel negative samples, features, and an ensemble classifier [J]. Artificial Intelligence in Medicine, 2017,83: 67-74.
[11] OLIVER S. Proteomics: guilt-by-association goes global [J]. Nature, 2000, 403(6770): 601-603.
[12] CHI X, HOU J. An iterative approach of protein function prediction [J]. BMC Bioinformatics, 2011, 12(1): 437-445.
[13] XIONG W, XIE L, GUAN J, et al. Active learning for protein function prediction in protein-protein interaction networks [C]// Proceedings of the 8th IAPR International Conference on Pattern Recognition in Bioinformatics. Berlin: Springer, 2014: 172-183.
[14] WANG H, HUANG H, DING C. Function-function correlated multi-label protein function prediction over interaction networks [C]// Proceedings of the 16th Annual International Conference on Research in Computational Molecular Biology. Berlin: Springer, 2012: 302-313.
[15] TENG Z, GUO M, LIU X, et al. Revealing protein functions based on relationships of interacting proteins and GO terms [J]. Journal of Computational Biology, 2013,20(4): 322-343.
[16] YU G, WANG J, LIU J. Protein function prediction by random walks on a hybrid graph [J]. Current Proteomics, 2016, 13(2): 130-142.
[17] HARTWELL L H, HOPFIELD J J, LEIBLER S, et al. From molecular to modular cell biology [J]. Nature, 1999, 402(6761 Suppl):47-52.
[18] RIVES A W, GALITSKI T. Modular organization of cellular networks [J]. Proceedings of the National Academy of Sciences of the United States of America, 2003, 100(3): 1128-1133.
[20] CLAUSET A, NEWMAN M E J, MOORE C. Finding community structure in very large networks [J]. Physical Review E: Statistical, Nonlinear, and Soft Matter Physics, 2004, 70(6): 066111.
[21] NEWMAN M E J, GIRVAN M. Finding and evaluating community structure in networks [J]. Physical Review E: Statistical, Nonlinear, and Soft Matter Physics, 2004, 69(2): 026113.
[22] ABDI H, WILLIAMS L J. Principal component analysis [J]. Wiley Interdisciplinary Reviews Computational Statistics, 2010, 2(4): 433-459.
[23] GILLIS J, PAVLIDIS P. The impact of multifunctional genes on “guilt by association” analysis [J]. PLOS ONE, 2011, 6(2): e17258.
[24] CARPENTER G A, GROSSBERG S. Self-organizing neural networks for supervised and unsupervised learning and prediction [M]// From Statistics to Neural Networks, NATO ASI Series 136. Berlin: Springer, 1994: 319-348.
[25] GLOROT X, BORDES A, BENGIO Y. Deep sparse rectifier neural networks [EB/OL]. [2017- 03- 01]. http://proceedings.mlr.press/v15/glorot11a/glorot11a.pdf.
[26] 劉威,劉尚,周璇.BP神經網絡子批量學習方法研究[J].智能
系統學報,2016,11(2):226-232.(LIU W, LIU S, ZHOU X. Subbatch learning method for BP neural networks [J]. CAAI Transactions on Intelligent Systems, 2016, 11(2):226-232.)
[27] XENARIOS I, RICE D W, SALWINSKI L, et al. DIP: the database of interacting proteins [J]. Nucleic Acids Research, 2000, 28(1): 289-291.
[28] ASHBURNER M, BALL C A, BLAKE J A, et al. Gene ontology: tool for the unification of biology [J]. Nature Genetics, 2000, 25(1): 25-29.
[29] MULDER N J, APWEILER R, ATTWOOD T K, et al. InterPro, progress and status in 2005 [J]. Nucleic Acids Research, 2005, 33(Database issue): D201-D205.
[30] CONSORTIUM U P. The Universal Protein resource (UniProt) in 2010 [J]. Nucleic Acids Research, 2010, 38(Database issue): 142-148.
[31] DURINCK S, SPELLMAN P T, BIRNEY E, et al. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt [J]. Nature Protocols, 2009, 4(8):1184-1191.
[32] RADIVOJAC P, CLARK W T, ORON T R, et al. A large-scale evaluation of computational protein function prediction [J]. Nature Methods, 2013, 10(3):221-227.
This work is partially supported by the National Natural Science Foundation of China (61363035, 61762015), the Natural Science Foundation of Guangxi (2015GXNSFAA139288), the “Bagui Scholars” Project, the Systematic Research Foundation of Guangxi Key Laboratory of Multi-source Information Mining and Safety (14-A-03-02, 15-A-03-02), the Guangxi Graduate Education Innovation Program (XYCSZ2017067).
TANGJiaqi, born in 1992, M. S. candidate. His research interests include bioinformatics, machine learning.
WUJingli, born in 1978, Ph. D., professor. Her research interests include bioinformatics, algorithm design and analysis.
猜你喜歡
中華詩詞(2022年6期)2022-12-31 06:41:24
瘋狂英語·新策略(2019年10期)2019-12-13 08:43:28
當代陜西(2019年10期)2019-06-03 10:12:04
數學小靈通·3-4年級(2017年9期)2017-10-13 08:10:54
中國科技論壇(2017年7期)2017-07-25 08:49:53
軟件導刊(2016年9期)2016-11-07 21:35:42
通信電源技術(2016年5期)2016-03-22 01:09:49
石油知識(2016年2期)2016-02-28 16:20:16
自動化儀表(2015年11期)2015-04-01 01:02:40
中國中醫藥現代遠程教育(2014年22期)2014-03-01 04:32:55
