磷脂酶A1輔助蛋白N端截短菌株的構建及其優化表達
2018-06-14 06:45:28朱昊薛正蓮王洲陳阿娜楊蒙
食品與發酵工業 2018年5期
關鍵詞:優化
朱昊,薛正蓮,2*,王洲,2,陳阿娜,2,楊蒙
1(安徽工程大學 生物與化學工程學院,安徽 蕪湖,241000)2(微生物發酵安徽省工程研究中心,安徽 蕪湖,241000)
磷脂酶A1是一類專一水解磷脂sn-1位酰基產生溶血磷脂和自由脂肪酸的酶類,廣泛存在于各種動植物和微生物體中[1]。磷脂酶A1生物功能可歸納為3類:細胞膜結構的維護和修復;細胞內代謝機制和信號傳導的調節及體內磷脂的消化[2]。 溶血磷脂可被廣泛應用于食品、化妝品、飼料改良和醫藥等領域[3]。
蘇燕南等[4]在研究粘質沙雷氏菌生產磷脂酶A1的過程中發現:在磷脂酶A1編碼基因plaA的下游存在一段輔助蛋白編碼序列plaS基因,它與磷脂酶A1在大腸桿菌中的高活性表達密切相關,同時顯示對宿主細胞有抑制作用。進一步的研究表明,此輔助蛋白PlaS屬于錨蛋白ANK家族[5]。ANK家族錨蛋白是一類廣泛存在于自然界的調節蛋白質相互作用的蛋白質,在細胞生理功能的實現和調控方面具有重要意義[6]。如在銅綠假單胞菌中存在一段類似于磷脂酶輔助蛋白的基因(ANKB),這段基因和過氧化酶基因屬于同一個操縱子中,且位于下游無重疊基因,屬于ANK家族,ANKB蛋白位于細胞周質空間,具有信號肽,對于過氧化氫酶的表達具有重要的調控作用[7]。ALCOFORADO等[8]的研究也發現在粘質沙雷氏菌中六型分泌表達系統和輔助蛋白對其種間競爭有一定的調節作用。
為進一步揭示粘質沙雷氏菌中磷脂酶A1輔助蛋白PlaS對磷脂酶A1合成及對宿主細胞生長行為調控的機制,深入研究磷脂酶A1輔助蛋白PlaS具有重要的理論和實踐指導意義。目前,國內外對磷脂酶A1輔助蛋白PlaS的相關研究報道較少。本實驗室張爽等[9]在前期實驗中發現輔助蛋白plaS基因在大腸桿菌表達系統中構建成功后很難表達,僅在Western Blot技術下才見有少量表達,而且對宿主菌的生長也有一定的抑制作用。國內外均有研究者通過截短氮端促進蛋白表達[10-13]。
本實驗去除了plaS基因的前35個氨基酸,使其與pET-28a(+)載體連接,轉化到大腸桿菌BL21(DE3)中,實現了目的蛋白的異源表達。通過對實驗條件的優化,使目的蛋白達到最大表達量,為輔助蛋白胞外可溶性表達奠定了基礎,也為輔助蛋白的純化及后續功能的深入研究提供了參考。
1 材料與方法
1.1 菌株、質粒與儀器
粘質沙雷氏菌PL-06,BL21(DE3),質粒載體pET-28a(+)為本實驗室保存,P28為本實驗室在前期實驗中將pET-28a(+)載體質粒導入BL21(DE3)表達宿主中構建的空載,SP28為本實驗室在前期實驗中以pET-28a(+)為載體,插入plaS基因片段,導入BL21(DE3)表達宿主中構建并保存的菌株。
本文使用寧波新芝牌超聲波細胞粉碎機對細胞進行破碎。
1.2 培養基與試劑
LB培養基(1 L):酵母膏5 g、蛋白胨10 g、NaCl 10 g、H2O 1 000 mL、pH 7.0。
卡那霉素(Kan)溶液:向8 mL滅菌去離子水中加入0.5 g卡那霉素,溶解后定容至10 mL。經0.22 μm濾膜過濾,分裝置于在-20 ℃保存。
100 mmol/L IPTG溶液:向40 mL滅菌去離子水中加入1.19 g 的IPTG,溶解后定容至50 mL。經0.22 μm濾膜過濾,分裝置于在-20 ℃保存。
LB+Kan固體培養基:待融化的LB固體培養基冷卻到一定溫度時, 加入卡那霉素,使其終質量濃度為50 μg/mL。
1%瓊脂糖凝膠:向20 mL 1×TAE緩沖液中加入0.2 g瓊脂糖粉,加熱使完全溶解,待冷卻至50~60 ℃時加入1 μL EB,混勻后倒入制膠模具中插上梳子,室溫冷卻凝固即可。
限制性核酸內切酶BamHⅠ和HindⅢ 購自賽默飛世爾科技公司。Taq酶購于BBI生物公司,其他試劑為國產分析純。
1.3 輔助蛋白N端截短菌株的設計和構建
通過對輔助蛋白的氨基酸序列預測與比對,發現PlaS輔助蛋白序列與錨定蛋白重組子家族(ANK家族)存在一定同源性,氨基酸序列在種間保守性較高,相似度達80%。對之前在NCBI上釋放的輔助蛋白PlaS序列進行信號肽分析,發現前35AA為信號肽區域(圖1)。
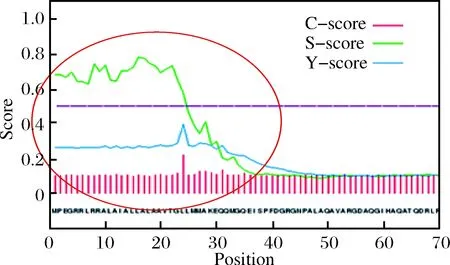
圖1 信號肽分析Fig.1 Signal peptide analysis
為使蛋白成功表達,綜合考慮后決定通過PCR方法去除前35AA。根據已知的plaS基因序列,用Primer 5.0軟件設計截斷N端的引物,引物序列見表1。
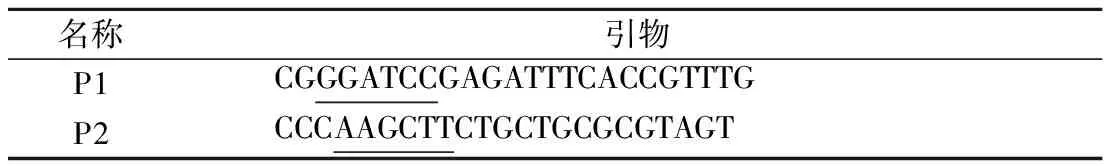
表1 dSP28引物序列Table 1 dSP28 Primers
注:劃線部分分別為BamHⅠ、HindⅢ酶切位點。
以本實驗室前期構建的SP28質粒為模板, P1,P2為引物,用Taq酶進行PCR擴增dplaS基因,擴增條件為:95 ℃ 5 min;95 ℃ 35 s;56 ℃ 35 s;72 ℃ 45 s;72 ℃ 8 min。對擴增后的基因片段進行膠回收,雙酶切后與pET-28a(+)載體鏈接,并轉化至BL21(DE3)感受態細胞中。隨后挑取陽性克隆子進行PCR驗證,并送上海生工生物有限公司測序,將測序正確的菌種進行保種,即得到1株構建成功的dSP28表達菌株。
1.4 dSP28誘導表達及條件優化
以P28空載為對照,從LB+Kan的平板上分別挑取P28和dSP28單菌落至100 mL LB+Kan的液體培養基中,37 ℃,200 r/min過夜培養,按2%的接種量接種至100mL LB+Kan的培養液中,至OD600nm值為0.7時加入0.2 mmol/L IPTG對其進行誘導,誘導4 h后取菌液制備蛋白樣品,并進行SDS-PAGE蛋白電泳,確定目的蛋白表達部位。根據已確定的蛋白表達部位,分別對誘導時間、IPTG濃度、初始OD600nm值和溫度進行優化,提高蛋白表達量。其中誘導時間分別為:2、4、6、8、10 h;IPTG誘導濃度分別為: 0、0.2、0.4、0.6、0.8、1.0 mmol/L;初始OD600nm值分別為: 0.1,0.2,0.4,0.7,0.9;溫度分別為: 28、31、34、37、40、43、46 ℃。
1.5 P28、SP28與dSP28在最優表達條件下OD600nm值的測定
從LB+Kan的平板上分別挑取P28、SP28和dSP28單菌落至100 mL LB+K的液體培養基中,37 ℃,200 r/min過夜培養,按2%的接種量接種至100 mL LB+Kan的培養液中,按最佳誘導條件進行培養,每2 h取樣測1次OD600nm值,并繪制發酵曲線。
1.6 SDS-PAGE蛋白電泳樣品制備
1.6.1 發酵液上清蛋白的制備
(1)取20 mL菌液,用離心機于12 000 r/min離心10 min,取18 mL上清液至干凈的離心管中;
(2)在4 ℃下,緩慢加入95%的乙醇18 mL;
(3)將二者充分振蕩,于-20 ℃放置10 min;
(4)4 ℃,12 000 r/min,離心15 min,去上清,晾干;
(5)加入400 μL TE緩沖液重懸沉淀;
(6)從重懸沉淀中取80 μL,加入20 μL 5×Loading buffer混合后,100 ℃煮5 min;
(7)于12 000 r/min離心1 min,取上清蛋白20 μL進行蛋白電泳。
1.6.2 全菌樣品的制備
取500 μL菌液,用離心機于9 000 r/min離心2 min,棄上清,加入80 μL TE,吹懸后加入20 μL 5×Loading buffer,100 ℃煮5 min,12 000 r/min,離心1 min,取上清蛋白20 μL進行蛋白電泳。
1.6.3 破碎上清蛋白的制備
(1)取20 mL發酵液,用離心機于4 000 r/min離心5 min,收集菌體沉淀;
(2)取20 mL PB緩沖液重懸菌體,于4 000 r/min離心5 min,收集菌體沉淀,重復此步驟1次;
(3)將收集到的沉淀用20 mL PB緩沖液重懸,冰浴10 min,用細胞破碎儀300 W,3 s工作,3 s停頓,超聲破碎10 min;
(4)將破碎后的菌液于12 000 r/min離心10 min,取18 mL上清液至干凈的離心管中;
(5)在4 ℃下,緩慢加入95%的乙醇18 mL;
(6)將二者充分振蕩數分鐘,于-20 ℃放置10 min;
(7)4 ℃,12 000 r/min,離心15 min,去上清,晾干;
(8)加入400 μL TE緩沖液重懸沉淀;
(9)從重懸沉淀中取80 μL,加入20 μL 5×Loading buffer混合后,100 ℃煮5 min;
(10)于12 000 r/min離心1 min,取上清蛋白20 μL進行蛋白電泳。
1.6.4 破碎菌體蛋白的制備
(1)用20 mL PB緩沖液重懸超聲破碎后離心得到的沉淀;
(2)從(1)中取500 μL重懸液,用離心機9 000 r/min,離心2 min,棄上清,加入80 μL TE,吹懸后加入20 μL 5×Loading buffer 100 ℃煮5 min,12 000 r/min,離心1 min,取上清蛋白20 μL進行蛋白電泳。
2 結果與分析
2.1 dSP28菌株的構建和驗證
以引物P1和P2擴增得到的片段大小為651 bp(圖2),將擴增好的片段與載體相連,轉化。隨后,從帶有Kan抗性的平板上挑取轉化子,接種至5 mL LB+Kan液體培養基中過夜培養,次日,從該菌液中提取質粒作為模板,以P1,P2為引物進行PCR驗證,根據瓊脂糖電泳圖中條帶大小判斷陽性克隆子(圖3),并將PCR驗證正確的菌株送上海生工生物有限公司測序,用DNAMAN對序列進行分析比對,序列一致則對該菌株保種,用于后續實驗。
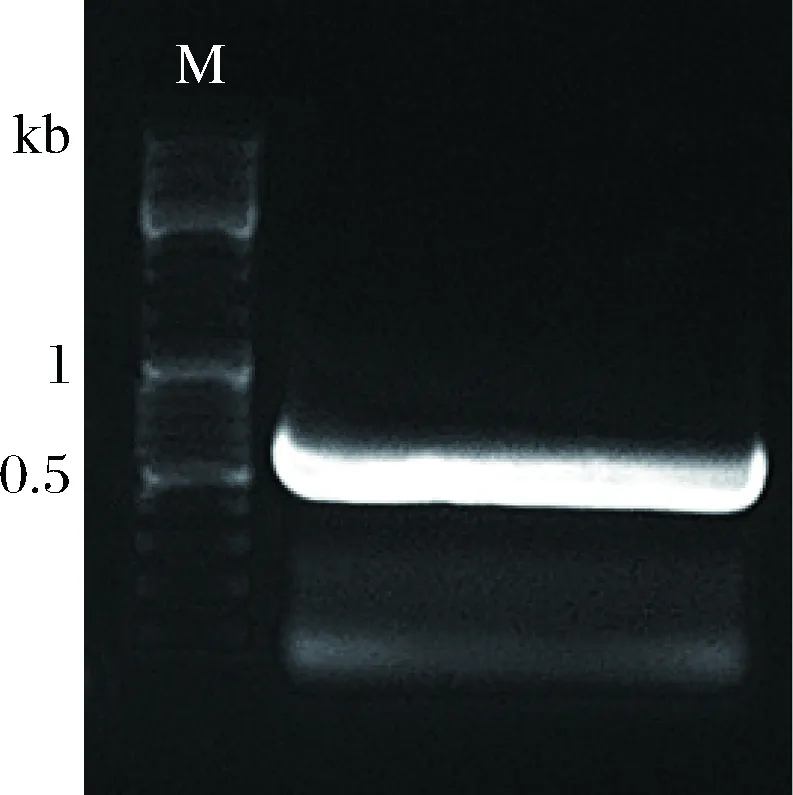
M-Marker圖2 SP28中dplaS的PCR擴增Fig.2 PCR amplification gene dplaS and in SP28
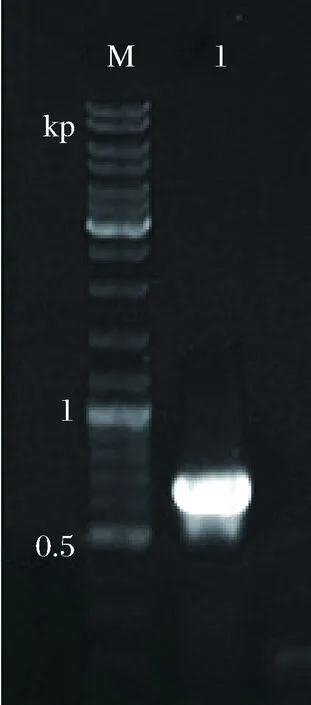
M-marker; 1-菌落PCR結果圖3 dSP28菌落PCR驗證Fig.3 The colony PCR of dSP28
2.2 dSP28目的蛋白表達位置判斷
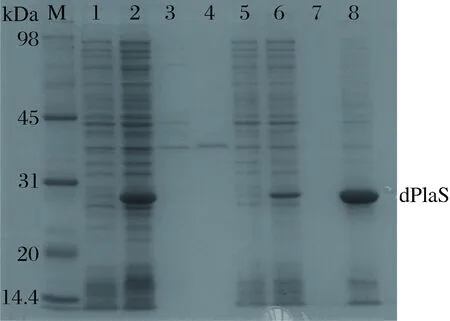
M-marker;1、3、5、7分別是P28的全菌、發酵液上清蛋白、破碎上清蛋白、破碎菌體蛋白;2、4、6、8分別是dSP28的全菌、發酵液上清蛋白、破碎上清蛋白、破碎菌體蛋白圖4 P28和dSP28SDS-PAGEFig.4 The SDS-PAGE of P28 and dSP28
按1.4的方法,分別對P28和dSP28進行誘導表達,制備蛋白樣品。SDS-PAGE蛋白電泳結果(圖4)表明,dSP28中目的蛋白在全菌和破碎菌體中均有較高的表達量,后續優化實驗可通過全菌中目的蛋白在電泳圖中相應分子量處條帶粗細變化來確定優化條件。
2.3 dSP28表達條件的優化
2.3.1 誘導時間的優化
以2%的接種量將dSP28種子液接種至100mL LB+Kan的液體培養基中,至OD600nm值為0.7時加入0.2 mmol/L IPTG,于37 ℃,200 r/min對其進行誘導,在誘導后2、4、6、8、10 h分別取樣制備蛋白電泳。結果(圖5)可知,最佳誘導時間為加入IPTG后8 h。
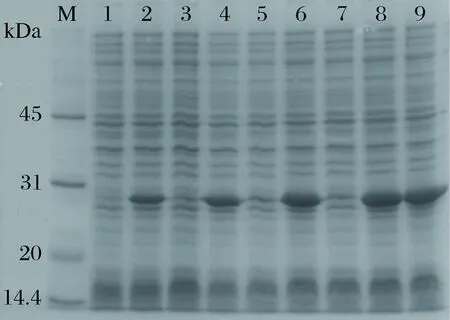
M-marker;1、3、5、7-P28的誘導時間分別為:2、4、6、8 h;2、4、6、8、9dSP28的誘導時間分別為:2、4、6、8、10 h圖5 誘導時間對dSP28表達的影響Fig.5 Effect of induction time on dSP28 protein expression
2.3.2 IPTG濃度的優化
以2%的接種量將dSP28種子液接種至100 mL LB+Kan的液體培養基中,至OD600nm值為0.7時,分別加入0、0.2、0.4、0.6、0.8、1.0 mmol/L的IPTG,37 ℃,200 r/min誘導8 h,制備蛋白樣品,進行電泳。通過SDS-PAGE蛋白電泳圖發現(圖6),最佳IPTG濃度為0.2 mmol/L。
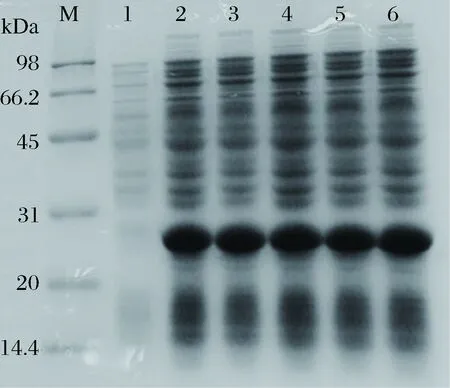
M-marker;1~6-IPTG 0、0.2、0.4、0.6、0.8、1.0 mmol/L圖6 IPTG濃度對dSP28表達的影響Fig.6 Effect of IPTG concentration on dSP28 protein expression
2.3.3 初始OD600nm值的優化
以2%的接種量將dSP28種子液接種至100 mL LB+Kan的液體培養基中,在初始OD600nm值分別為: 0.1、0.2、0.4、0.7、0.9時,加入0.2 mmol/L IPTG,37 °C,200 r/min誘導8 h,制備蛋白樣品。SDS-PAGE蛋白圖電泳表明(圖7),最佳初始OD600nm值為0.7。
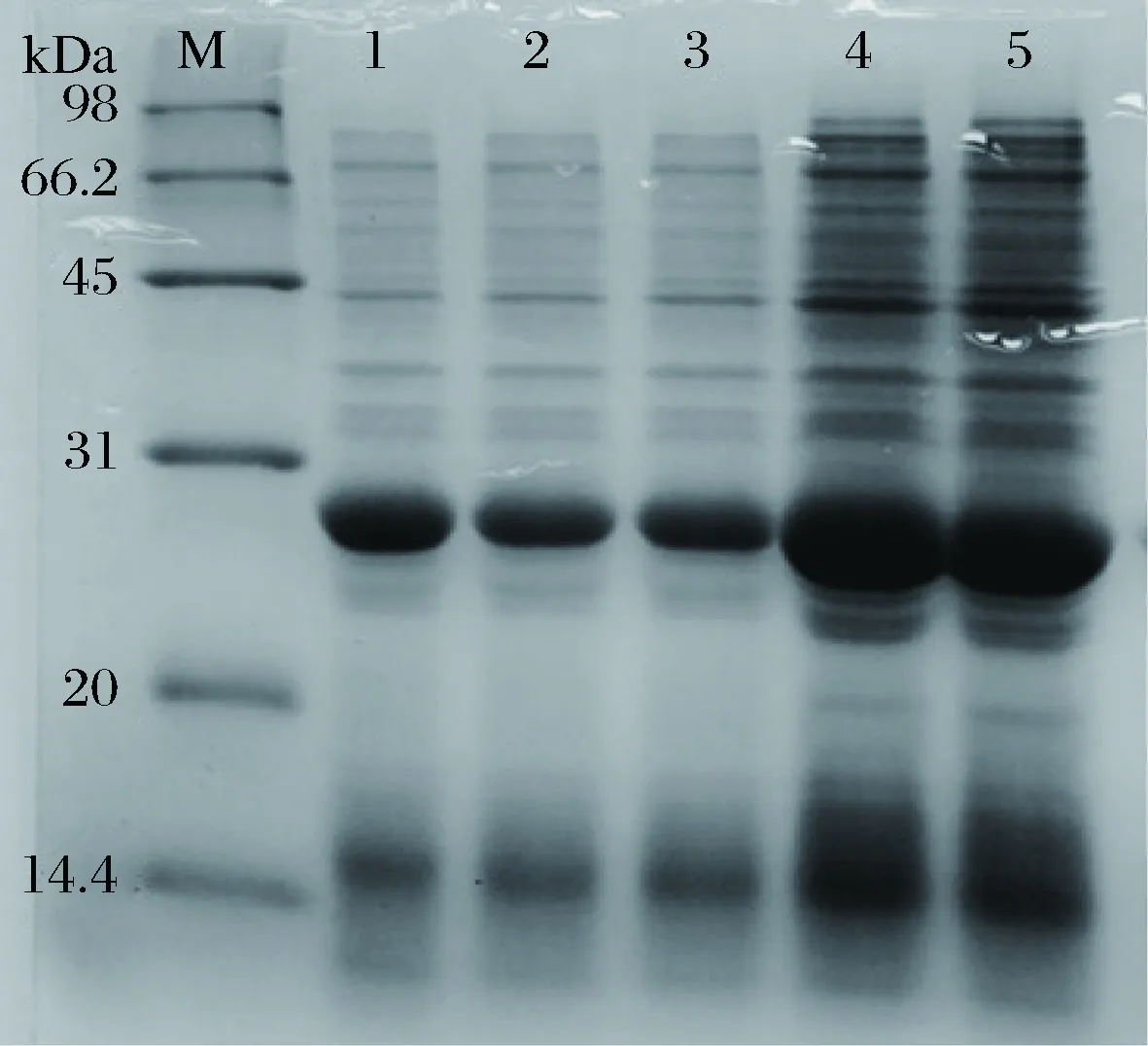
M-marker;1~5-OD600nm值分別為:0.1、0.2、0.4、0.7、0.9圖7 初始OD600nm值對dSP28表達的影響Fig.7 Effect of induction starting OD600nm on dSP28 protein expression
2.3.4 誘導溫度的優化
以2%的接種量將dSP28種子液接種至100 mL LB+Kan的液體培養基中,在初始OD600nm值為0.7時,加入0.2 mmol/L IPTG,將誘導溫度梯度設置為:28、31、34、37、40、43、46 ℃,誘導8 h,制備蛋白樣品。結果發現(圖8),最佳誘導溫度為40 ℃。
M:marker;1~7-28、31、34、37、40、43、46 ℃圖8 溫度對dSP28表達的影響Fig.8 Effect of induction temperature on dSP28 protein expression
2.4 P28、SP28和dSP28發酵中OD600nm值的測定
經過表達條件的優化,得到dSP28中dPlaS蛋白的最佳表達條件:誘導時間為8 h,IPTG濃度為0.2 mmol/L,起始菌濃(OD600nm值)為0.7,溫度為40 ℃,轉速為200 r/min。以此表達條件對P28、dSP28和SP28誘導前后OD600nm值進行測定(圖9),發現在按2%接種量接種后, 37 ℃,200 r/min,每0.5 h測1次OD600nm值時,3者菌濃差異并不明顯。而加入IPTG開始誘導后,40 ℃,200 r/min,每2 h測1次OD600nm值時,SP28的菌濃先增長后降低,并趨于平穩,同時顯示輔助蛋白PlaS對宿主細胞有較強的生長抑制作用。P28和dSP28則一直處于增長趨勢,且增幅較大,直到終止誘導,其依舊處于上升狀態。這一現象說明輔助蛋白N端截短對PlaS蛋白表達有一定的促進作用,且生長較快,對宿主菌的抑制作用大大減弱,為后面實驗奠定了基礎。
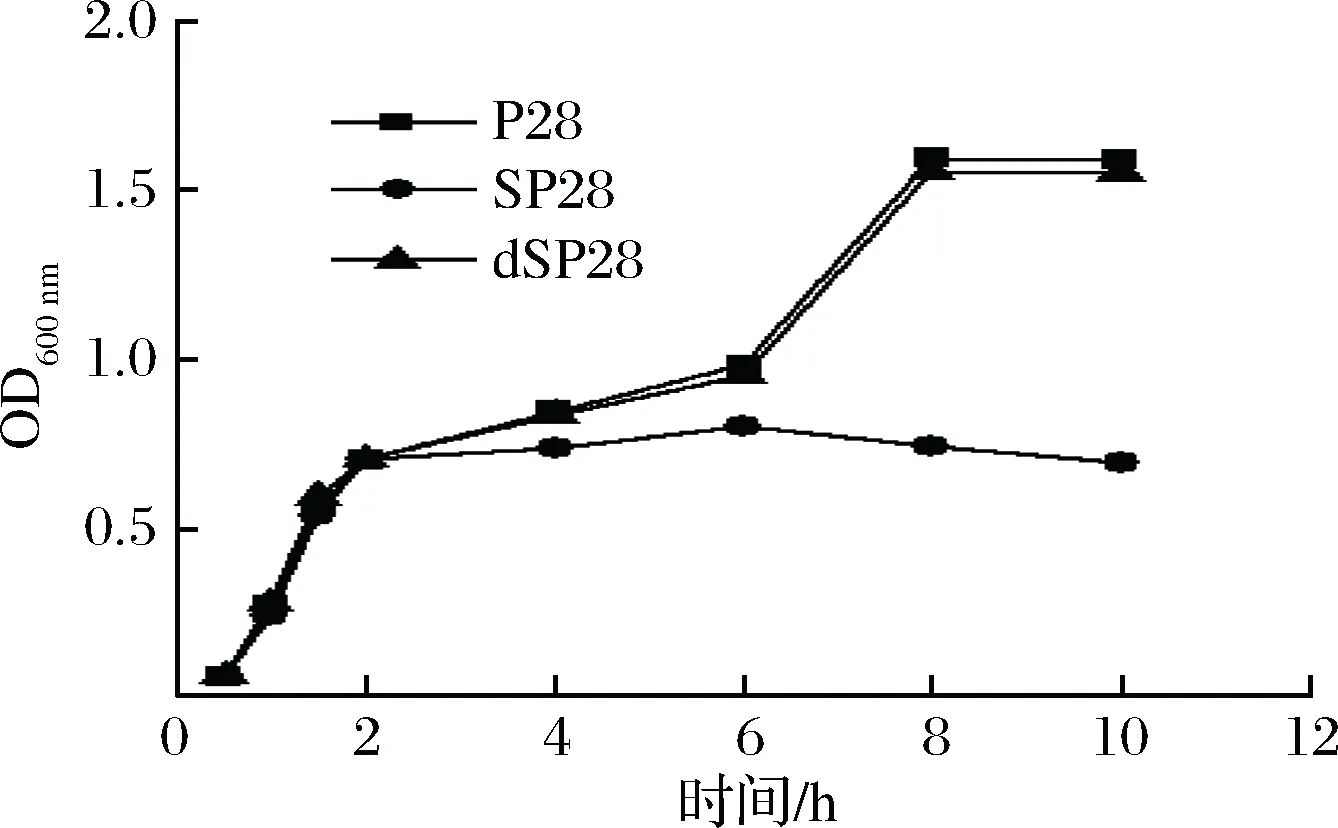
圖9 P28、SP28和dSP28發酵中OD600nm值測定Fig.9 Determination of OD600nm in P28, SP28 and dSP28 fermentation
3 討論
張爽等[9]在前期實驗研究中發現,磷脂酶A1輔助蛋白dPlaS在大腸桿菌表達系統中構建成功后表達較為困難,只在Western Blot技術下有極少量的目的蛋白表達條帶,且對構建的工程菌生長有一定的抑制作用,這可能與其信號肽有關。在本實驗中對plaS基因N端截短,去除了信號肽后發現,輔助蛋白可以在細胞內進行大量表達,進行表達條件優化后,在誘導時間為8 h,IPTG濃度為0.2 mmol/L,起始菌濃(OD600nm)為0.7,溫度為40 ℃,轉速為200 r/min時在全菌中達到了最大表達量,而通過發酵中OD600nm值測定,發現其在最優表達條件下對宿主菌生長抑制作用明顯減弱。這為后期蛋白大量胞外表達和純化奠定了基礎,也為進一步研究輔助蛋白對磷脂酶A1酶活的影響,及對輔助蛋白功能特性的深入研究提供了可能。
[1] RICHMOND G S, SMITH T K.Phospholipases A1[J].Mol Sci, 2011, 12(1):588-612.
[2] VICTOR C, DIANA M, CARLOS T, et al.Phospholipases in food industry: a review [J].Methods Mol Biol, 2012(861): 495-523.
[3] 王利.大豆磷脂酶水解技術及磷脂的HLB值測定[D].長春:中國人民解放軍軍需大學, 2002.
[4] 蘇燕南.磷脂酶A1基因的克隆和原核表達[D].蕪湖:安徽工程大學,2013.
[5] 薛正蓮, 蘇燕南, 王洲,等.粘質沙雷氏菌錨定蛋白重復子及其應用: CN, CN 103333229 A[P].2013.
[6] AL-KHODOR S, PRICE C T, KALIA A, et al.Functional diversity of ankyrin repeats in microbial proteins[J].Trends in Microbiology, 2010, 18(3):132-139.
[7] HOWELL M L, ALSABBAGH E, MA J F, et al.AnkB, a periplasmicankyrin-like protein inPseudomonasaeruginosa, is required for optimal catalase B (KatB) activity and resistance to hydrogen peroxide[J].Journal of Bacteriology, 2000, 182(16):4 545-4 556.
[8] ALCOFORADO D J, COULTHURST S J.Intraspecies competition inSerratiamarcescensis mediated by type VI-secreted Rhs effectors and a conserved effector-associated accessory protein[J].Journal of Bacteriology, 2015, 197(14):2 350-2 360.
[9] 張爽.粘質沙雷氏菌磷脂酶A1輔助蛋白PlaS功能特性研究[D].蕪湖:安徽工程大學,2015.
[10] 程蓓蓓, 陳舜,汪銘書,等.四川白鵝CD4基因胞外去信號肽區的可溶性表達及純化[J].四川動物,2015(6):875-879.
[11] 王明道, 聶慧慧, 王紅陽,等.普魯蘭酶氮端氨基酸的截短對其酶學性質的影響[J].農業生物技術學報, 2016, 24(7):1 101-1 108.
[12] DUAN X, WU J.Enhancing the secretion efficiency and thermostability ofBacillusderamificanspullulanase mutant D437H/D503Y byN-terminal domain truncation[J].Applied & Environmental Microbiology, 2015, 81(6):1 926-1 931.
[13] GOPAL G J, KUMAR A.Strategies for the production of recombinant protein inEscherichiacoli[J].The Protein Journal, 2013, 32(6):1-17.
猜你喜歡
房地產導刊(2022年5期)2022-06-01 06:20:14
能源工程(2022年1期)2022-03-29 01:06:28
建材發展導向(2021年12期)2021-07-22 08:06:48
建材發展導向(2021年7期)2021-07-16 07:07:52
中學生數理化(高中版.高二數學)(2021年12期)2021-04-26 07:43:48
中學生數理化(高中版.高考數學)(2021年12期)2021-03-08 01:28:50
今日農業(2020年16期)2020-12-14 15:04:59
消費導刊(2018年8期)2018-05-25 13:20:08
家庭影院技術(2018年4期)2018-05-09 07:07:41
電子制作(2017年20期)2017-04-26 06:57:45
