基于HPLC波長切換法對芪歸通便合劑中六種成分的含量測定
2018-06-29 02:13:06陶健
實用藥物與臨床 2018年5期
陶 健
0 引言
芪歸通便合劑是我院的醫院制劑,由炙黃芪、酒肉蓯蓉、白術、升麻等十味中藥材提取加工而成。方中黃芪、白術補肺健脾,當歸補血活血為君藥;肉桂溫通血脈,肉蓯蓉、制首烏滋補肝腎、調和氣血、潤腸通便為臣藥;佐以炒決明子、炒火麻仁潤腸通便,升麻、陳皮調理氣機、健脾通便。諸藥合用,共奏益氣養血、滋補肝腎、潤腸通便的功效。中藥復方制劑成分復雜,單一成分難以控制產品質量的穩定性和均一性,為確保產品批次間的穩定和臨床用藥的安全有效,本文采用HPLC波長切換法對芪歸通便合劑中主藥黃芪所含主要成分毛蕊異黃酮葡萄糖苷和芒柄花素,白術所含主要成分白術內酯Ⅲ和白術內酯Ⅰ,酒肉蓯蓉所含主要成分松果菊苷和毛蕊花糖苷進行同時測定研究,填補了芪歸通便合劑質量標準在高效液相色譜法研究的空白。
1 儀器與試藥
HP-1100型高效液相色譜儀(美國安捷倫科技有限公司);AB104-N型電子天平(南京庚辰科學儀器有限公司)。芪歸通便合劑來源于平頂山市中醫醫院(每瓶裝500 mL,批號:20161216、20170120、20170216、20170307、20170401、20170426、20170509、20170529、20170612、20170627);松果菊苷對照品(82854-37-3)來源于上海純優生物科技有限公司;毛蕊花糖苷對照品(111530-201512)、毛蕊異黃酮葡萄糖苷對照品(111920-201606)、芒柄花素對照品(111703-201504)、白術內酯Ⅲ對照品(111978-201501)和白術內酯Ⅰ對照品(111975-201501)均來源于中國食品藥品檢定研究院;甲醇和乙腈為色譜純,甲酸為分析純。
2 方法與結果
2.1 色譜條件 色譜柱:Diamonsil C18柱(250 mm×4.6 mm,5 μm);流速:0.8 mL/min;柱溫:30 ℃;進樣量為10 μL;流動相A:甲醇-乙腈(1∶1),流動相B:0.1%甲酸溶液,梯度洗脫(0~10 min,23.0%A;10~18 min,23.0%A→30.0%A;18~27 min,30.0%A→42.0%A;27~35 min,42.0%A→65.0%A;35~40 min,65.0%A→23.0%A);0~18 min時在330 nm[1-2]波長下檢測松果菊苷和毛蕊花糖苷,18~27 min在254 nm[3]波長下檢測毛蕊異黃酮葡萄糖苷和芒柄花素,27~40 min在220 nm[4-5]波長下檢測白術內酯Ⅲ和白術內酯Ⅰ。在此系統條件下,理論塔板數按所測色譜峰計均不低于3 500,分離度大于1.5。
2.2 溶液的制備
2.2.1 混合對照品溶液 精密稱取對照品松果菊苷、毛蕊花糖苷、毛蕊異黃酮葡萄糖苷、芒柄花素、白術內酯Ⅲ、白術內酯Ⅰ各適量,分別用甲醇溶解并制成單一成分的對照品儲備液(各成分質量濃度分別為松果菊苷2.418 mg/mL、毛蕊花糖苷1.102 mg/mL、毛蕊異黃酮葡萄糖苷0.510 mg/mL、芒柄花素0.788 mg/mL、白術內酯Ⅲ 0.416 mg/mL、白術內酯Ⅰ 0.382 mg/mL);依次精密吸取對照品儲備液松果菊苷2.5 mL、毛蕊花糖苷2.5 mL、毛蕊異黃酮葡萄糖苷1.0 mL、芒柄花素2.5 mL、白術內酯Ⅲ 2.5 mL、白術內酯Ⅰ 2.5 mL,置同一50 mL量瓶中,用甲醇稀釋至刻度,制成測定用混合對照品溶液(所測各成分質量濃度分別為松果菊苷0.120 9 mg/mL、毛蕊花糖苷0.055 1 mg/mL、毛蕊異黃酮葡萄糖苷0.010 2 mg/mL、芒柄花素0.039 4 mg/mL、白術內酯Ⅲ 0.020 8 mg/mL、白術內酯Ⅰ 0.019 1 mg/mL)。
2.2.2 供試品溶液和陰性對照溶液的制備 精密吸取芪歸通便合劑3.0 mL,置25 mL量瓶中,用甲醇稀釋至刻度,搖勻,濾過,取續濾液作為芪歸通便合劑供試品溶液。依據芪歸通便合劑處方和制備工藝,分別制備不含炙黃芪的陰性樣品、不含酒肉蓯蓉的陰性樣品和不含白術的陰性樣品,再按照芪歸通便合劑供試品溶液制備方法,分別制成黃芪陰性對照溶液、肉蓯蓉陰性對照溶液和白術陰性對照溶液。
2.3 方法學驗證
2.3.1 線性關系考察 分別精密吸取松果菊苷、毛蕊花糖苷、毛蕊異黃酮葡萄糖苷、芒柄花素、白術內酯Ⅲ、白術內酯Ⅰ對照品儲備液0.1、0.2、0.5、1.0、1.5、2.0 mL,置于6個20 mL容量瓶中,分別用甲醇稀釋至刻度,制成6個濃度梯度的混合對照品溶液,依法進樣測定,以質量濃度C為橫坐標,松果菊苷、毛蕊花糖苷、毛蕊異黃酮葡萄糖苷、芒柄花素、白術內酯Ⅲ、白術內酯Ⅰ的峰面積A為縱坐標進行線性回歸,回歸方程、線性范圍和相關系數結果見表1。
2.3.2 專屬性實驗 分別精密吸取黃芪陰性對照溶液、肉蓯蓉陰性對照溶液、白術陰性對照溶液、混合對照品溶液和芪歸通便合劑供試品溶液各適量,依法進樣測定,結果表明,松果菊苷、毛蕊花糖苷、毛蕊異黃酮葡萄糖苷、芒柄花素、白術內酯Ⅲ、白術內酯Ⅰ相互測定均無干擾,且陰性對6個成分的測定無干擾,色譜圖見圖1。

表1 6個成分的回歸方程和線性范圍

圖1 混合對照品(A)、芪歸通便合劑(B)、肉蓯蓉陰性(C)、黃芪陰性(D)、白術陰性(E)色譜圖
2.3.3 精密度試驗 取混合對照品溶液,重復進樣6次,記錄松果菊苷、毛蕊花糖苷、毛蕊異黃酮葡萄糖苷、芒柄花素、白術內酯Ⅲ、白術內酯Ⅰ的峰面積,結果所測6個組分峰面積的RSD依次為0.95%、1.08%、1.36%、1.03%、0.79%、1.45%。
2.3.4 重復性試驗 取芪歸通便合劑(批號:20161216)6份,按照供試品溶液制備過程分別制成重復性試驗供試品溶液,依法進樣檢測,分別計算松果菊苷、毛蕊花糖苷、毛蕊異黃酮葡萄糖苷、芒柄花素、白術內酯Ⅲ、白術內酯Ⅰ的含量,并計算所測6個組分含量的RSD,結果所測6個組分含量的RSD依次為1.22%、0.37%、1.79%、0.53%、1.34%、1.65%。
2.3.5 加樣回收試驗 取已知含量的芪歸通便合劑(批號:20161216)適量,精密吸取9份,每份1.5 mL,分別置25 mL量瓶中,依據中國藥典2015年版四部《藥品質量標準分析方法驗證指導原則》,分別加入0.834 mg/mL松果菊苷對照品溶液1.0 mL 3份、2.0 mL 3份、3.0 mL 3份,0.387 mg/mL毛蕊花糖苷對照品溶液1.0 mL 3份、2.0 mL 3份、3.0 mL 3份,0.132 mg/mL毛蕊異黃酮葡萄糖苷對照品溶液0.5 mL 3份、1.0 mL 3份、1.5 mL 3份,0.421 mg/mL芒柄花素對照品溶液0.5 mL 3份、1.0 mL 3份、1.5 mL 3份,0.292 mg/mL白術內酯Ⅲ對照品溶液0.5 mL 3份、1.0 mL 3份、1.5 mL 3份,0.214 mg/mL白術內酯Ⅰ對照品溶液0.5 mL 3份、1.0 mL 3份、1.5 mL 3份,按照芪歸通便合劑供試品溶液制備方法制成高、中、低3種濃度,每種濃度3份的加樣回收樣品溶液,依法進樣檢測,計算這6個組分的平均加樣回收率及RSD值,結果見表2。
2.4 樣品含量測定 取批號分別為20161216、20170120、20170216的芪歸通便合劑,按“2.2.2”項下的供試品溶液制備方法制備供試品溶液,依法進樣檢測松果菊苷、毛蕊花糖苷、毛蕊異黃酮葡萄糖苷、芒柄花素、白術內酯Ⅲ和白術內酯Ⅰ的含量,結果見表3。
3 討論
在流動相的選擇時,本實驗首先考察對比了甲醇-水體系[5]、乙腈-水體系、甲醇-乙腈(1∶1)與水體系[6],結果甲醇-乙腈(1∶1)與水體系所測各成分的分離效果優于甲醇-水體系和乙腈-水體系,但所測成分松果菊苷、毛蕊花糖苷峰形欠佳,存在拖尾現象;在此基礎上,對比考察了甲醇-乙腈(1∶1)與0.1%甲酸溶液體系[1-3]、甲醇-乙腈(1∶1)與0.2%磷酸溶液[4,7-8]、甲醇-乙腈(1∶1)與1%醋酸溶液體系[9-10],以色譜峰基線的平穩情況、所測各成分的峰形和分離度為綜合考察指標,最終確定采用甲醇-乙腈(1∶1)與0.2%磷酸溶液為流動相,同時對流動相的比例進行摸索,最終確定了采用“2.1”項下的色譜條件對芪歸通便合劑中松果菊苷、毛蕊花糖苷、毛蕊異黃酮葡萄糖苷、芒柄花素、白術內酯Ⅲ和白術內酯Ⅰ進行同時測定。
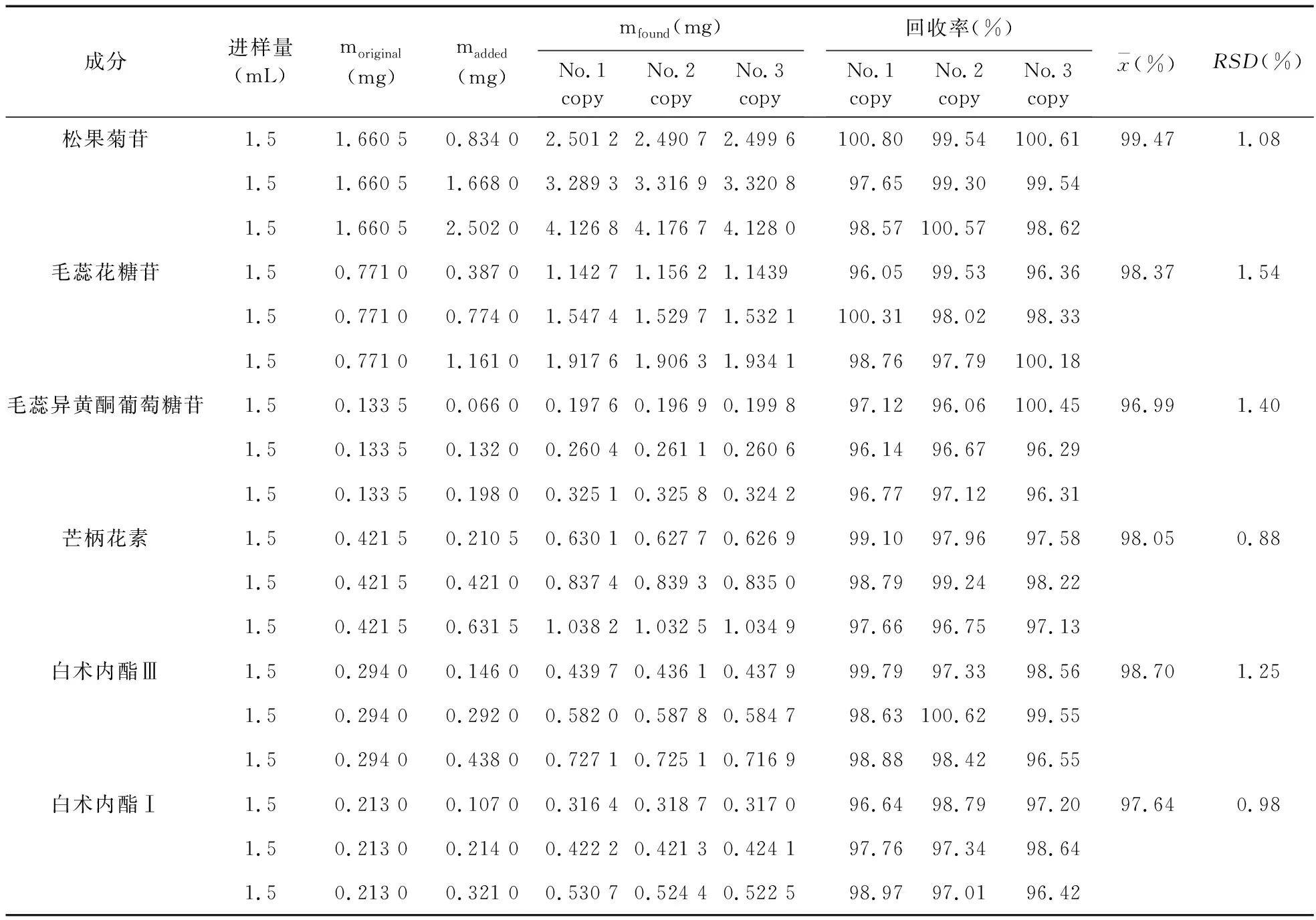
表2 松果菊苷、毛蕊花糖苷、毛蕊異黃酮葡萄糖苷、芒柄花素、白術內酯Ⅲ和白術內酯Ⅰ回收率

表3 含量測定結果(n=3,mg/mL)
從試驗結果來看,在相同的生產工藝和測定方法下,不同批次芪歸通便合劑所含松果菊苷、毛蕊花糖苷、毛蕊異黃酮葡萄糖苷、芒柄花素、白術內酯Ⅲ和白術內酯Ⅰ存在一定的差異,可能與不同批次芪歸通便合劑所用中藥材中相關成分含量的差異有關,因此,建立芪歸通便合劑中多成分同時測定方法,并對所用中藥材制定符合該制劑的內控質量標準勢在必行,從而有效保證芪歸通便合劑質量的穩定性,確保臨床用藥的安全有效。根據10個批次芪歸通便合劑所測各成分含量平均值的80%,暫定芪歸通便合劑每1 mL含酒肉蓯蓉以松果菊苷計不得少于0.85 mg,以毛蕊花糖苷計不得少于0.40 mg;含炙黃芪以毛蕊異黃酮葡萄糖苷計不得少于0.07 mg,以芒柄花素計不得少于0.23 mg;含白術以白術內酯Ⅲ計不得少于0.16 mg,以白術內酯Ⅰ計不得少于0.11 mg。本實驗采用波長切換法同時測定芪歸通便合劑中6種成分的含量,方法靈敏度高,檢測操作方法簡便,對控制芪歸通便合劑產品質量的均一性和療效一致性提供了理論基礎及數據支持。
參考文獻:
[1] 國家藥典委員會.中華人民共和國藥典(一部)[S] .北京:中國醫藥科技出版社,2015:103-104,135,302-303.
[2] 田玉彪,楊廣安,黃維杰,等.HPLC測定肉蓯蓉微粉中松果菊苷和毛蕊花糖苷的含量[J] .安徽農業科學,2015,43(33):171-173.
[3] 付娟,楊世海,黃林芳.超高效液相色譜法同時測定黃芪中6種黃酮類成分的含量[J] .中國藥學雜志,2013,48(11):916-919.
[4] 劉青青,金傳山,吳德玲,等.不同加工工藝對白術中白術內酯Ⅰ和白術內酯Ⅲ含量的影響[J] .安徽中醫學院學報,2012,31(1):58-61.
[5] 許祥君,陳敏,宗露,等.不同品規白術中白術內酯Ⅰ、Ⅲ的含量測定[J] .中國醫院藥學雜志,2011,31(17):1421-1424.
[6] 紀松崗,李翔,婁子洋,等.高效液相色譜法測定黃芪中毛蕊異黃酮苷和芒柄花素的含量[J] .第二軍醫大學學報,2006,27(1):81-84.
[7] 王波,周圍,劉小花,等.基于超高效合相色譜對黃芪中5種主要黃酮類化合物的快速檢測[J] .分析化學,2016,44(5):731-739.
[8] 李志浩,瞿京紅,劉菁,等.高效液相色譜法測定固本咳喘片中白術內酯Ⅰ和白術內酯Ⅲ的含量[J] .湖北中醫藥大學學報,2011,13(4):33-35.
[9] 張思巨,劉麗,于江泳,等.HPLC同時測定肉蓯蓉藥材中松果菊苷和毛蕊花糖苷的含量[J] .中國藥學雜志,2004,39(10):740-741.
[10] 陳敏,肖蘇萍,崔光紅,等.管花肉蓯蓉中松果菊苷和毛蕊花糖苷的含量測定[J] .中國中藥雜志,2005,30(11):839-841.
