血清脂肪細胞型脂肪酸結合蛋白和白細胞介素-17水平與2型糖尿病患者下肢動脈粥樣硬化病變的相關性研究
2018-07-04 11:02:54鄭亞虹許慕蓉萬麗娟陳明衛
安徽醫科大學學報 2018年7期
劉 瑋,鄭亞虹,沈 穎,許慕蓉,萬麗娟,陳明衛
糖尿病下肢動脈粥樣硬化病變(Lower extremity atherosclerotic disease,LEAD)是糖尿病最常見的慢性并發癥之一。在我國根據踝肱指數(ankle-brachinal index,ABI)檢查,50歲以上患者中LEAD患病率高達6.9%~23.8%。LEAD的發病機制較為復雜,目前公認的病理機制包括內皮功能的損害、氧化應激等。近年來,免疫炎癥在LEAD發病中的作用日益受到重視[1]。脂肪細胞型脂肪酸結合蛋白(adiopocyte fatty acid binding protein,A-FABP)體內分布廣泛,主要是在脂肪細胞和巨噬細胞中表達,在調節脂肪酸的轉運、巨噬細胞轉化為泡沫細胞以及炎癥因子的釋放過程發揮重要作用[2],與肥胖和代謝綜合征密切相關。白細胞介素-17(interleukin-17, IL-17)主要表達于外周記憶性CD4+T細胞以及血管內皮細胞、嗜酸性粒細胞等,是一種新近發現的強大的促炎細胞因子[3]。目前已有報道A-FABP、IL-17與動脈粥樣硬化疾病密切相關[4-5],但對2型糖尿病(type 2 diabetes mellitus, T2DM)合并LEAD中的A-FABP、IL-17濃度變化尚不清楚。該研究通過檢測T2DM合并LEAD患者血清A-FABP、IL-17水平改變,分析A-FABP、IL-17與LEAD發病之間的相關性,以期為T2DM合并LEAD的臨床防治提供指導。
1 材料與方法
1.1病例資料選取2015年10月~2016年8月在安徽醫科大學第一附屬醫院內分泌科住院的臨床診斷為T2DM的患者129例,其中男74例,女55例,年齡39~75(58.82±13.33)歲。根據ABI檢查結果將入選的129例患者分為無下肢動脈粥樣硬化病變組(NLEAD組,n=90)(0.9≤ ABI<1.3)和合并下肢動脈粥樣硬化病變組(LEAD組,n=39)(ABI<0.9)。另選擇性別、年齡匹配的40例健康人群作為對照組 (NC組),均來自于同一時間來本院進行健康體檢人群。所有研究對象均無急慢性感染性疾病、惡性腫瘤、肝功能異常以及自身免疫性疾病。本研究經安徽醫科大學第一附屬醫院醫學倫理委員會批準,并獲取受試者知情同意。
1.2研究方法
1.2.1資料收集 收集患者性別、年齡、病程,測量收縮壓(SBP)、舒張壓(DBP)、身高、體質量,計算體質量指數(BMI)=體質量/身高2(kg /m2) 。
1.2.2生化指標檢測 所有受試者經10 h的空腹過夜,抽肘靜脈血測定血總膽固醇(TC)、甘油三酯(TG)、高密度脂蛋白膽固醇(HDL-c)、低密度脂蛋白膽固醇(LDL-c)、空腹血糖(FBG)、糖化血紅蛋白(HbA1c)、空腹C肽(FCP)、血肌酐(SCr)、尿素氮(BUN)水平。采用李霞 等[6]方法根據FCP及FBG,用穩態模型計算胰島素抵抗[HOMA-IR (CP)=1.5+FBG(mmol/L)×FCP(pmol/L)/2 800]及胰島功能[HOMA-islet (CP)=0.27×FCP(pmol/L)/(FBG(mmol/L)-3.5)+50]。
1.2.3ABI的測量 患者取平臥位,將袖帶分別綁扎于上臂及踝上,采用日本ES-1000spm多普勒血流探測儀探頭測量足背、脛后及肱動脈收縮壓值,肱動脈收縮壓取雙側肱動脈收縮壓的高值,一側踝動脈收縮壓取足背與脛后動脈收縮壓的高值,一側踝動脈收縮壓與肱動脈收縮壓的比值即為該側的ABI。
1.2.4血清A-FABP、IL-17的測定 清晨空腹抽取研究對象靜脈血5 ml,調整離心機至2 500 r/min、4 ℃的情況下,對樣本進行10 min的離心,所得血清存放在-80 ℃的冰箱中保存待測。檢測前放置-4 ℃的冰箱中解凍30 min,通過酶聯免疫測定法(ELISA)分別測定血清A-FABP、IL-17的濃度。操作過程嚴格參照試劑盒說明書。
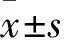
2 結果
2.1三組間一般臨床資料與血清A-FABP、IL-17水平的比較三組間性別、年齡、BMI、DBP、TC、TG、HDL-c、BUN比較,差異無統計學意義;NLEAD組與LEAD組間在FCP、HOMA-IR、HOMA-islet方面比較,差異無統計學意義。與NC組比較,NLEAD組、LEAD組的SBP、LDL-c、HbA1C、A-FABP、IL-17濃度均顯著升高(P<0.05)。此外,LEAD組中SCr也顯著高于NC組(P<0.05),而NLEAD組血Cr與NC組則差異無統計學意義;與NLEAD組比較,LEAD組中病程、SBP、LDL-c、血Cr、HbA1c、FBG、A-FABP、IL-17濃度均顯著增高(P<0.05)。見表1。
2.2A-FABP、IL-17與其他因素的相關分析Pearson相關分析顯示,A-FABP與BMI、HbA1c、SBP、HOMA-IR、Scr、IL-17呈正相關,r值分別為0.224、0.164、0.147、0.183、0.173、0.203,P值分別為0.008、0.031、0.048、0.017、0.024、0.011;而A-FABP與年齡、病程、TC、TG、HDL-c、FCP、FBG、BUN、HOMA-islet均無明顯相關性。IL-17與LDL-c、HbA1c、SBP呈正相關,r值分別為0.162、0.255、0.152,P值分別為0.047、0.001、0.042;與HOMA-islet呈負相關,r值為-0.204,P值為0.013;而IL-17與年齡、病程、BMI、TC、TG、HDL-c、FCP、FBG、BUN、Cr、HOMA-IR均無明顯相關性。

表1 三組間一般情況及生化指標的比較
與NC組比較:*P<0.05;與NLEAD組比較:#P<0.05;P1值:三組間比較;P2值:NLEAD組與LEAD組比較
2.3LEAD的Logistic逐步回歸分析在T2DM患者中(包括LEAD組及NLEAD組),在單因素分析基礎上,以有無LEAD為應變量,單因素分析P<0.1的變量為候選變量(包括年齡、病程、SBP、BMI、HbA1c、LDL-c、Cr、HOMA-islet、A-FABP、IL-17)基礎上,進行Logistic回歸分析,結果顯示LDL-c、A-FABP、IL-17為 LEAD發病的獨立危險因素。見表2。

表2 LEAD危險因素的Logistic回歸分析
3 討論
近年研究顯示,多種脂肪因子、炎癥因子的水平改變與T2DM及其慢性并發癥的發生發展密切相關。本研究顯示,T2DM組血清A-FABP、IL-17水平較NC組均顯著升高,相關分析顯示,A-FABP與胰島素抵抗呈正相關,IL-17與胰島功能指數呈負相關關系,提示A-FABP、IL-17均可能參與T2DM發病過程。然而在最近發表的一些研究中,卻沒有發現T2DM與健康對照人群中A-FABP、IL-17水平存在顯著差異[7-8]。可能與種族差異、研究對象選擇不同等因素有關。
有研究[9]顯示T2DM合并大血管病變患者血清IL-17水平明顯增高,但該研究納入的大血管病變除了LEAD以外,還包括心腦血管疾病,因此對T2DM合并LEAD血清IL-17水平的變化仍不清楚。目前關于T2DM合并LEAD患者血清A-FABP變化的研究甚少。最近發表的一項研究[10]顯示,T2DM合并周圍血管病變患者血清A-FABP水平較無周圍血管病變患者顯著增高,并與女性患者周圍血管病變發病關系更加密切。本研究顯示,LEAD組A-FABP、IL-17水平均顯著高于NLEAD組,Logistic回歸分析表明,A-FABP、IL-17均是影響LEAD的獨立危險因素,可使LEAD的發病風險分別增加1.945倍和2.372倍,提示T2DM患者中,A-FABP、IL-17與LEAD的發生發展密切相關。此外,相關分析顯示,A-FABP 與BMI、HbA1c、SBP、HOMA-IR均呈正相關;IL-17與LDL-c、HbA1c、SBP均呈正相關關系;A-FABP 與IL-17呈正相關,提示A-FABP、 IL-17作為脂肪因子/促炎細胞因子,彼此之間可能存在交互影響,并可通過與多種心血管危險因素相互作用,參與到LEAD發病進程中[5,10]。
目前有關A-FABP、IL-17參與LEAD的發生、發展的具體機制尚未闡明。目前研究顯示,動脈粥樣硬化是血脂紊亂與炎癥反應共同參與的慢性疾病。巨噬細胞中A-FABP過表達,可使三酰甘油和膽固醇沉積,形成泡沫細胞,促進動脈粥樣硬化的形成,此效應可能與A-FABP調控腫瘤壞死因子α、巨噬細胞炎癥蛋白-1α、單核細胞趨化蛋白-1等炎癥因子、化學趨化因子產生與炎癥反應有關[11]。在肥胖小鼠體內A-FABP基因敲除可以對抗動脈粥樣硬化,改善胰島素抵抗及脂代謝異常。IL-17與IL-17受體結合后,調節免疫應答,可誘導白介素-6、腫瘤壞死因子-α、趨化因子等的表達,產生致炎作用,促進炎癥進程[3]。在T2DM大鼠模型中,主動脈內膜中IL-17 mRNA及蛋白表達增加,IL-17可參與血管內膜損傷以及大血管病變的炎癥反應過程[12]。體外實驗發現,IL-17能上調或協同基質金屬蛋白酶-9、γ-干擾素、C反應蛋白表達[13-15]。
導致LEAD發生的危險因素眾多,目前公認的危險因素主要包括血糖、血壓、吸煙、糖尿病病程、脂代謝紊亂、慢性腎臟病等[16]。在本組資料中LEAD組的SBP、LDL-c、Cr、HbA1c、FBG水平均較NLEAD組增高,Logistic回歸分析表明,LDL-c是影響LEAD的獨立危險因素,表明嚴格控制血壓、血脂及血糖,預防慢性腎臟病發生對降低LEAD發生風險有積極的作用。
總之,本研究顯示,T2DM合并LEAD患者外周血A-FABP、IL-17水平明顯增加,A-FABP、IL-17可能參與LEAD的發病過程,具體的致病機制有待進一步研究闡明。
[1] Signorelli S S,Katsiki N. Oxidative stress and inflammation: their role in the pathogenesis of peripheral artery disease with or without type 2 diabetes mellitus[J]. Curr Vasc Pharmacol,2017,doi:10.2174/1570161115666170731165121.
[2] Furuhashi M, Tuncman G, G?rgün C Z, et al.Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2[J]. Nature, 2007, 447 (7147): 959-65.
[3] Kim B S,Park Y J,Chung Y.Targeting IL-17 in autoimmunity and inflammation [J].Arch Pharm Res, 2016,39(11):1537-47.
[4] Autieri M V. Adipose inflammation at the heart of vascular disease[J]. Clin Sci (Lond), 2016, 130 (22): 2101-4.
[5] Lu X. The Impact of IL-17 in atherosclerosis[J]. Curr Med Chem,2017,24 (21):2345-58.
[6] 李 霞,周智廣,亓海英,等. 用空腹C肽代替胰島素改良HOMA公式評價胰島素抵抗和胰島B細胞功能[J].中南大學學報(醫學類),2004,29(4):419-23.
[7] 張 珊,謝新苗,楊梅麗,等. 血清Irisin水平與脂肪細胞型脂肪酸結合蛋白的相關性研究[J]. 中國糖尿病雜志,2017,25(7):594-8.
[8] Qiao Y C, Jian S, Lan H, et al. Changes of regulatory T cells and of proinflammatory and immunosuppressive cytokines in patients with type 2 diabetes mellitus: a systematic review and meta-analysis[J]. J Diabetes Res, 2016,2016:3694957.
[9] 樓旭丹,王海東,陳 芳,等. 2型糖尿病大血管病變患者血清IL-17水平變化及意義[J]. 中國老年學,2013,33(13):3024-6.
[10] Ding M, Shi J Y, Xing Y Z, et al. Serum adipocyte fatty acid-binding protein levels are associated with peripheral arterial disease in women, but not men, with type 2 diabetes mellitus [J]. J Diabetes, 2017,doi:10.1111//1753-0407.12549.
[11] Wu G, Li H, Zhou M, et al. Mechanism and clinical evidence of lipocalin-2 and adipocyte fatty acid-binding protein linking obesity and atherosclerosis[J]. Diabetes Metab Res Rev, 2014,30(6):447-56.
[12] Erbel C, Akhavanpoor M, Okuyucu D, et al. IL-17A influences essential functions of the monocyte/macrophage lineage and is involved in advanced murine and human atherosclerosis[J]. J Immunol, 2014,193(9):4344-55.
[13] Cheng G, Wei L, Wang X, et al. IL-17 stimulates migration of carotid artery vascular smooth muscle cells in an MMP-9 dependent manner via p38 MAPK and ERK1/2-dependent NF-κB and AP-1 activation[J]. Cell Mol Neurobiol, 2009, 29(8):1161-8.
[14] Onishi R M, Gaffen S L. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease[J]. Insect Sci, 2010, 129(3):311-21.
[15] Patel D N, King C A, Bailey S R, et al. Interleukin-17 stimulates C-reactive protein expression in hepatocytes and smooth muscle cells via p38 MAPK and ERK1/2-dependent NF-κB and C/EBPβ activation[J]. J Biol Chem, 2007, 282(37):27229-38.
[16] Rhee S Y, Kim Y S. Peripheral arterial disease in patients with type 2 diabetes mellitus[J]. Diabetes Metab J, 2015, 39 (4):283-90.
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
中老年保健(2021年3期)2021-08-22 06:50:04
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
天津醫科大學學報(2021年2期)2021-03-29 05:31:08
現代臨床醫學(2021年1期)2021-01-26 00:56:02
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
人大建設(2019年12期)2019-05-21 02:55:32
現代檢驗醫學雜志(2014年4期)2014-02-02 02:44:59
