B、N共摻雜單層石墨烯電子結構和導電性能
2018-07-23 02:22:54任福成徐守冬劉世斌
太原理工大學學報 2018年4期
關鍵詞:結構
任福成,徐守冬,張 鼎,陳 良,劉世斌
(太原理工大學 潔凈化工研究所,太原 030024)
近年來鋰電池在生活中得到了很廣泛的應用[1-4],但鋰電池的能量密度尚不能達到汽油的能量密度(13 000 Wh/kg)[5]。為了使鋰電池在未來能夠應用在運輸工具上,對鋰電池的研究重點集中到發展新型鋰電池體系,比如Li-O2[6-14]和Li-S[15-16]電池等。在這些新型電池中,Li-O2電池能量密度可達到11 682 Wh/kg,和化石燃料相當,因此具有很好的應用前景。Li-O2電池的高能量密度由電池負極金屬鋰和正極O2反應得到,O2可直接從空氣中分離獲取。然而Li-O2[17-18]電池的發展還處在初級階段,推廣應用受很多因素的限制,如電解液的穩定性不高、較高的反應過電勢、低放電容量和循環性能差[19-21]等。提高電池的反應性能的常用方法之一就是采用電化學催化劑,如基碳類材料[22-23]、貴金屬[24-25]、金屬氧化物[26-27]和合金類催化劑[28]等;其中,基碳類催化劑因其大比表面積、高導電性和低成本等而受到廣泛重視。在基碳類材料中,石墨烯因其質量密度輕(2 g/cm3)、比表面積大(2 630 m2/g)、化學穩定性高和金屬性質的導電性而被廣泛運用到Li-O2電池的正極材料中。KIM et al[29]將石墨烯作為負極材料,電池循環第1周和第10周的庫倫效率分別為99%和87%.WU et al[30]合成的N摻雜石墨烯納米片應用到Li-O2電池中,對ORR反應的催化性能與Pt/C催化劑相當。ZHAO et al[31]合成的3D多孔石墨烯氣溶膠作為正極材料,在3.2 A·g-1下得到高比容量(5 978 mAh·g-1)和長循環壽命。
第一性原理研究方法在研究材料性能和反應機理方面有很大優勢。JING et al[32]計算5種不同結構類型的N摻雜石墨烯,表明平面結構N摻雜石墨烯對ORR催化活性最好。REN et al[33]的研究表明,B摻雜石墨烯對鋰電池中的析氧反應(OER)有促進作用,電池的充電速率明顯提高。N摻雜石墨烯可作為ORR反應的一種高效催化劑,但是N摻雜比例和位置會不同程度地影響其帶隙值。石墨氮摻雜的導電性隨摻N量的增加而降低,而吡啶氮摻雜石墨烯(PNG)的導電性能隨摻N量的增加先提高后降低[34]。LAREF et al[35]對不同結構類型的石墨烯分別進行B、N單元素摻雜,結果表明,B原子摻雜增強了石墨烯的導電性能,而N原子摻雜后其導電性能降低且隨摻雜比例增加而減弱。ZHAO et al[36]的研究認為,B、N原子間位共摻雜活化了C原子的pz軌道電子,使石墨烯對ORR、OER反應有很好的催化作用;然而在Li原子存在的環境中,其催化效果并不明顯。
電極材料的性能直接影響電池的應用。B、N單元素摻雜石墨烯作為具有催化性能的電極材料,研究者在其性能方面做了大量的研究,但是對摻雜后石墨烯的電荷分布、電子軌道雜化和軌道分布等研究很少。利用密度泛函理論研究B、N共摻雜石墨烯電子結構分布、導電性能以及共摻雜石墨烯材料在電池中的應用是很有必要的。本文對B、N原子以鄰、間、對位共摻雜石墨烯的電子結構進行詳細研究。B、N原子共同摻雜到一個碳環中,共摻雜模型和本征石墨烯是等電子體系,B、N共摻雜結構的電荷分布有明顯的轉移,共摻雜結構的能帶和原子間的雜化類型較石墨烯發生了很大的變化,本文通過對能帶、態密度和電子分布的分析來預測摻雜對材料性能影響。
1 計算方法和晶胞模型
1.1 計算方法
本次計算采用美國Accelerys公司開發的MS6.0(Materials Studio)軟件中的CASTEP模塊。幾何優化和電子結構計算基于密度泛函理論(DFT)平面波贗勢展開,采用廣義梯度近似(GGA)修正的PBE泛函處理交換關聯能[37-38],用超軟贗勢來描述價電子和離子實之間的相互作用。所有的計算都是在倒易空間中進行,幾何優化過程中的算法都采用BFGS。本文計算平面波截斷能(Energy cut-off)取為380 eV,第一布里淵的K-point設置為4×4×2,幾何優化參數設置如下:迭代過程中的SCF收斂精度為2.0×10-5eV/atom,最大位移為2.0×10-4nm,原子間相互作用收斂標準設定為2.0×10-5eV/atom,內應力不超過0.1 GPa.為減小層與層之間的相互影響,真空度取2.5 nm.計算過程中的價電子組態分別為B:1s22s22p1,C:1s22s22p2,N:1s22s22p3.
1.2 晶胞模型
本文分別對晶胞模型為2×2×1石墨烯和B、N原子以鄰、間、對位共摻雜石墨烯電子結構做詳細研究,石墨烯結構如圖1(a)所示。石墨烯結構中每個碳環由6個碳原子構成,每個碳原子被3個碳六環共用,所以石墨烯元胞有2個碳原子。圖1(b)-圖1(d)分別為B、N以鄰、間、對位共摻雜石墨烯的結構示意圖。B、N共摻雜石墨烯結構幾何對稱性降低,但3種共摻雜結構中B、C、N的原子比均為1∶6∶1(12.5%),B和N原子以相同比例摻雜石墨烯,保證摻雜結構和石墨烯是等電子體系,在等電子體系下研究B、N共摻雜對電子結構的影響,N摻雜量為7.54%和9%石墨烯對電池的循環和倍率性能有明顯的改善[39-40]。3種結構的形成能分別為:鄰位-2.31 eV,對位-1.20 eV,間位-1.14 eV。這一規律和JIANG et al[41]的結果一致,而且B、N鄰位摻雜結構比石墨烯的二維結構還要穩定,所以這幾種結構在反應過程中是穩定的[42-43]。
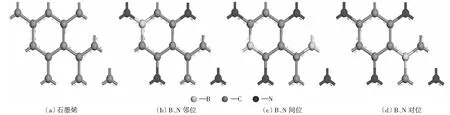
圖1 共摻雜2×2×1石墨烯的結構模型圖Fig.1 Schematic structure (top-view) of 2×2×1 graphene, B-NBG, S-NBG, P-BNG
2 結果與分析
2.1 能帶結構
為討論B和N以鄰位(B-BNG)、間位(S-BNG)、對位(P-BNG)共摻雜對單層石墨烯的電子結構的影響,本次研究分別對單層石墨烯和共摻雜單層石墨烯的能帶、態密度、電荷密度和電荷差分密度進行計算,能帶結構如圖2(a)-圖2(d)所示。從石墨烯的能帶結構分布(圖2(a))看出,在費米能級附近價帶最高點和導帶最低點,在布里淵區H-K點之間相切可知,其能帶分布具有金屬性,有半金屬性的導電性質[35]。如圖 2(b)-圖2(d)所示,B、N共摻雜對石墨烯的能帶分布產生很大影響,價帶和導帶之間出現帶隙,但都屬于直接帶隙。摻雜鄰、間、對位的帶隙值分別為2.406,1.296,2.572 eV;其中間位帶隙最小,而且間位共摻雜結構的能帶在布里淵區高對稱點H-K(圖2(c)),價帶和導帶同時出現最高點和最低點,與石墨烯相同。鄰位和對位的帶隙值接近,而且都在G點價帶和導帶發生分裂,如圖2(b)和圖2(d)所示,共摻雜形成的直接帶隙半導體材料中間位共摻雜結構的帶隙值最小,具有最好的導電性能。石墨N(GNG)摻雜比例為2.8%時,石墨烯能帶就出現帶隙,而且隨著摻雜比例升高帶隙增大,而且吡啶氮和吡咯氮對其帶隙值得影響也不同[34]。

圖2 共摻雜2×2×1石墨烯能帶結構圖Fig.2 Band structure of 2×2×1 graphene, B-NBG, S-NBG, P-BNG
共摻雜結構價帶的能帶不再簡單的分布,能帶增多而且能帶之間的交叉更為復雜,表現為B、N共摻雜改變了石墨烯結構中原子間的雜化類型,摻雜后的結構中存在C—C,B—C,C—N和B—C—N等共同雜化。摻雜后導帶的空軌道整體集中在2.5~7.5 eV,和石墨烯相比整體下移。B、N鄰位、間位、對位共摻雜能帶結構和石墨烯相比較的共同特點:價帶能帶數明顯增多,而且更靠近費米能級,能帶之間的交叉更為復雜,每條能帶所占的能量區間減小,所有的能帶結構較本征石墨烯變得平緩,電子軌道的擴展性減弱。LAREF et al[35]研究結果顯示:B摻雜石墨烯形成P型石墨烯,價帶能分布靠近費米能級,而N摻雜可得到N型石墨烯,導帶能分布靠近費米能級,同樣隨著摻雜比例升高石墨烯能帶結構中出現帶隙。通過對共摻雜后特殊的能帶結構分析可知,共摻雜結構在不同的環境下可作為很好的電子供體和受體。
2.2 態密度
圖3(a)-圖3(d)分別為石墨烯和B、N鄰位、間位、對位共摻石墨烯態密度圖。石墨烯態密度圖(圖3(a))費米能級兩側的峰,分別出現在-1.916,1.723 eV,主要來源于C原子2p軌道雜化的作用,兩峰之間的能量差為3.639 eV.B、N原子共摻雜石墨烯的態密度發生明顯變化,鄰位共摻雜費米能級兩側的第一個峰分別出現在-0.579,2.287 eV;從圖3(b)可以看出這兩個峰屬于B、C和N等3種原子的2p軌道共同雜化峰,能量差為2.866 eV.間位共摻雜費米能級兩側的第一個峰分別出現在-0.759,2.011 eV,雜化峰之間的能量差為2.760 eV,比鄰位共摻雜石墨烯的能量差小0.106 eV;其中,費米能級左側的第一個峰屬于B、C原子2p軌道的雜化峰,費米能級右側的第一個峰屬于B、C、N三原子的共同雜化峰,而且B、C原子間的雜化作用更強。對位費米能級兩側的第一個峰分別出現在-0.208,2.472 eV,兩個雜化峰之間的能量差為2.680 eV,比石墨烯的能量差小0.458 eV;其中,費米能級左側的第一個峰屬于B、C原子2p軌道的雜化峰,共摻雜結構在費米能級兩側態密度分布與石墨烯相比更靠近費米能級。N摻雜石墨烯其態密度在靠近費米能級的導帶向低能級移動,B摻雜石墨烯則相反價帶向高能級移動。計算結果表明,B和N共摻雜結構的價電子軌道和空軌道同時靠近費米能級,這一結果很好地結合了B和N單元素摻雜的特點[32-35]。對比圖3(a)-圖3(d)中費米能級兩側第一個峰,間位摻雜態密度的峰值和峰寬最大,表明共摻雜中間位可提供更多發生躍遷的電子,同時導帶底可為外電子提供更多空軌道。共摻雜結構中C—N原子雜化軌道分布在低能量區域,B—C原子雜化軌道分布更靠近費米能級,在-20 eV附近均存在一個C—N原子2s軌道的雜化峰。通過對能帶結構和態密度的分析可知,B、N共摻雜中間位共摻雜結構的帶隙最小,導電性能最優,化學活性也最強。
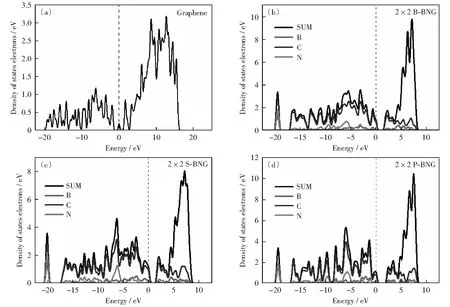
圖3 石墨烯,B、N鄰位,B、N間位,B、N對位共摻雜2×2×1石墨烯態密度圖Fig.3 Density of states of 2×2×1 graphene, B-NBG, S-NBG, P-BNG
2.3 間位共摻雜結構原子電荷布局分析
石墨烯的二維結構中所有C原子之間為sp2雜化,在C原子的2s和2p軌道分別分布1.05e和2.95e電子,pz軌道的電子形成π電子對。由于B、C、N等3種原子之間電負性的差異:B(2.04e),C(2.55e),N(3.04e),B和N原子間位取代兩個C原子后體系中N原子顯負電性,B原子顯正電性,C原子電荷分布平衡被打破,電荷分布發生偏移。從原子電荷布局分析(表1)可知,與B、N原子都成鍵的C2原子,從B原子得到的電子數比C2貢獻給N原子電子多0.1e;體系電荷重新分布后,B原子失去0.55e電子,比鄰位摻雜失去電子數少0.16e;2s和2p軌道上分別分布0.54e和1.91e電子,2p軌道上的電子在與C2,C4,C5的成鍵中起主要作用。與B原子成鍵的C5原子表現出最大的負電性,其2s和2p軌道分別得到0.09e和0.16e電子,N原子2s和2p軌道上分別分布1.41e和3.80e電子,得到0.21e電子。與N原子成鍵的C6原子,表現出最大的正電性,其2s軌道失去0.01e電子,2p軌道失去0.12e電子,二維結構中與B、N原子距離越大,C原子的電荷轉移數越小。通過間位共摻雜各原子2s和2p軌道電荷轉移情況的分析可知,與結構中B原子對C原子電荷轉移的影響相比,N原子的影響更為明顯;各原子的電荷轉移主要發生在原子的2p軌道,其原因是2p為B,C,N原子最外層電子軌道,B,N原子取代石墨烯結構中的C原子最先影響分布在原子最外層軌道上的電子。對比B,N鄰、間、對位共摻雜結構中C原子電荷轉移量可知,間位摻雜結構中C原子的電荷轉移量均小于鄰和對位摻雜,其中B原子的電荷轉移量由小到大為:間位(0.55e),對位(0.61e),鄰位(0.71e);N原子的電荷轉移量由小到大為:間位(-0.21e),對位(-0.25e),鄰位(-0.43e).
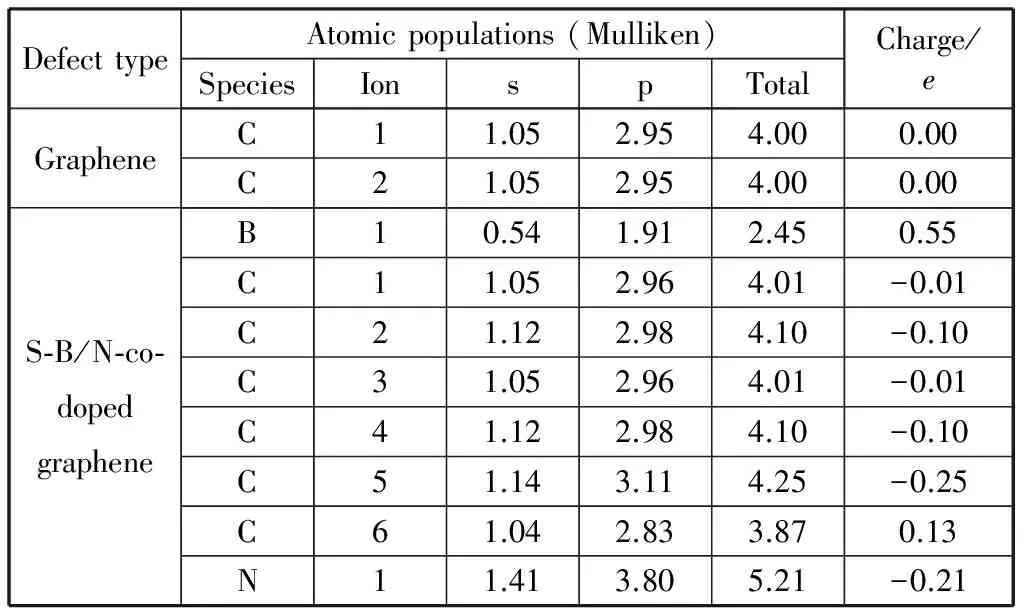
表1 石墨烯和B、N間位共摻雜2×2×1石墨烯原子電荷布局分析Table 1 Atom Mulliken-poppulation analysis of 2×2×1 Graphene and S-BNG
2.4 間位共摻雜結構電荷密度和電荷差分密度

圖4 石墨烯和B、N間位共摻雜石墨烯電荷密度圖Fig.4 Electron density contour of graphene and S-NBG
圖5為B、N間位共摻雜石墨烯的電荷差分密度圖,圖中藍色區域和紅色區域分別表示電荷密度減小(Δρ<0)和增大(Δρ>0).電荷差分密度Δρ計算公式:
Δρ=ρTotal-ρGraphene-∑nρi.
式中:ρTotal是摻雜體系優化后的總電荷密度分布函數;ρGraphene為石墨烯體系的電荷密度分布函數;ρi為B、N原子的電荷密度分布函數;n為B、N原子的摻雜數。在圖中的電荷密度值為0.017 4~0.716 8的區間選取了兩個電荷密度相等的面,其密度數值分別為0.273,0.443 .從等電荷密度面的分布可以看出,摻雜后電子云發生偏移,π電子對被破壞,等密度面不再沿著六邊形均勻分布,發生很大偏移。電荷密度為0.273的等密度面在B和C原子之間的偏移量很小,但是在C和N原子之間發生了明顯的偏移,更加靠近N原子;0.443的等密度面在B和C原子之間發生了偏移,偏離B原子靠近C原子,同樣,在C和N原子之間的分布也更靠近于N原子。在沒有和B、N原子成鍵的C原子之間,兩個等勢面均勻地分布在兩個C原子之間[45],摻雜之后材料的極性由非極性向極性轉變。

圖5 B、N間位摻雜石墨烯電荷差分密度圖Fig.5 Electron density difference of S-BNG
2.5 HOMO軌道
圖6為B、N原子間位共摻雜石墨烯的HOMO軌道(最高分子占據軌道)分布圖,能量范圍為-1.541 4~0.000 0 eV.理論上,HOMO電子的軌道和LUMO軌道(最低未占分子軌道)的能量差最小,即電子吸收能量躍遷到導帶空軌道的能量差最小。從圖6可以看出,最高占據態軌道主要分布在B、C原子周圍,N原子周圍只有很小一部分。B、N共摻雜2×2×1石墨烯結構的表面被活化,B和C原子的價電子更容易發生躍遷。
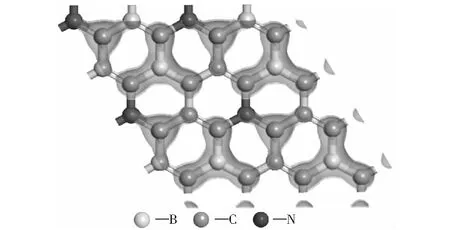
圖6 B、N間位共摻雜石墨烯HOMO軌道分布圖Fig.6 HOMO distribution of S-BNG
3 結論
本文利用密度泛函理論計算研究了B、N原子以鄰、間、對等3種位置共摻雜石墨烯結構的電子結構,詳細分析了B、N原子摻雜對石墨烯電子分布和導電性能的影響,以及B、C和N等3種原子之間的互相雜化關系,得出以下的結論。
1) 在B、N摻雜比例同為12.5%的條件下,共摻雜結構的能帶出現帶隙,都屬于直接帶隙;其中,間位的帶隙值最小(1.296 eV),而且間位摻雜結構和石墨烯的能帶結構中價帶最高點和導帶最低點相同,間位導電性能最好。
2) B、N共摻雜石墨烯的價電子軌道和空軌道分布更靠近費米能級,且B,C,N等3原子之間互相發生雜化和共同雜化,其雜化主要是各原子的2p軌道之間的雜化作用。其中,B、C原子價電子雜化軌道所占據的能量較高,靠近費米能級;B、C和N原子的價電子雜化軌道分布在低能量狀態;而C和N原子的空軌道分布更靠近費米能級。
3) B、N間位共摻雜結構中各原子的電荷轉移量最小,對石墨烯電子結構的影響最小,B原子失去0.55e電子,N原子得到0.21e電子。共摻雜結構表面被活化,分布在B和C原子周圍的價電子更容易發生躍遷,而N原子所在區域更容易接收外來電子。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
