轉錄因子碳水化合物反應元件結合蛋白研究進展
2018-08-29 02:59:28張明捷陳寒蓓
中國療養醫學 2018年8期
張明捷 陳寒蓓
Yamashita等[1]于2001年發現了一種與碳水化合物反應元件(carbohydrate response element,ChoRE)結合的蛋白質,將其命名為碳水化合物反應元件結合蛋白(carbohydrate response element binding protein,ChREBP)。ChREBP在哺乳動物的各種組織中廣泛表達,在肝臟、脂肪組織、小腸、肌肉和腎等組織中有較高的表達[2]。ChREBP是葡萄糖信號途徑中的轉錄因子,它直接激活多個參與糖酵解和脂肪合成基因的表達,對于調控體內糖、脂肪代謝具有十分重要的作用。
1 ChREBP的分子生物學特性
參與肝臟糖酵解及脂質合成的多種酶受碳水化合物的調控。葡萄糖能夠刺激肝臟丙酮酸激酶(liver pyruvate kinase,LPK)和脂肪酸合成酶(fatty acid synthase,FAS)等糖脂代謝相關酶的基因轉錄,從而促進葡萄糖轉化為脂肪。最初的研究發現,LPK等葡萄糖反應性基因的啟動子區存在著由間隔5bp的2個E盒(CAC GGG和CCC GTG)組成的ChoRE,其介導了葡萄糖對靶基因的轉錄激活作用[3]。2001年,Yamashita等首先分離純化到與LPK啟動子區ChoRE特異性結合的蛋白質,將其命名為ChREBP。
ChREBP屬于堿性螺旋-環-螺旋-亮氨酸拉鏈(basic helix-loop-helic/leucine zipper,bHLH-ZIP)轉錄因子家族,能夠識別靶基因中的Ebox序列。大鼠來源的ChREBP蛋白由864個氨基酸殘基組成,相對分子質量為946 000。ChREBP蛋白序列在不同類哺乳動物組織中具有高度同源性,人、大鼠和小鼠來源的ChREBP有82%的同源性[4]。ChREBP蛋白包含有多個不同的結構域,主要包含以下結構域[5]:N末端的核定位信號(nuclear localization signal,NLS)、C末端的b/HLH/Zip結構域、富含脯氨酸結構域(proline-rich domain,Pro-rich)和亮氨酸鋅指樣結構域(leucine-zipper-like domain,Zip-like)。此外,還含有4個與其活性密切相關的磷酸化位點:cAMP依賴蛋白激酶(cAMP-dependent protein kinase,PKA)和AMP激活蛋白激酶(AMP-activated protein kinase,AMPK)的磷酸化位點(圖1)。
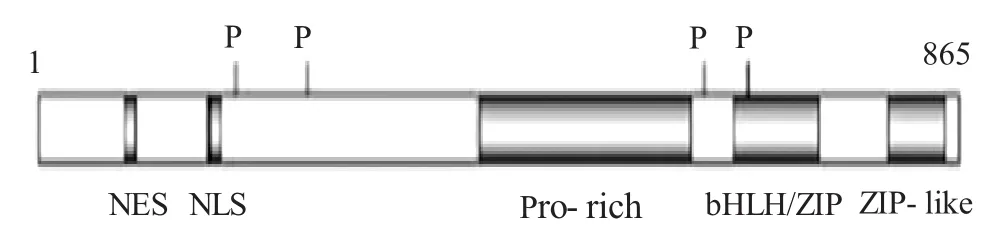
圖1 ChREBP結構圖[6]
2012年,Herman及其同事發現了一種ChREBP的新型異構體,稱為ChREBP-β[7],作者從1a起始位點翻譯產生了全長864個氨基酸的蛋白(ChREBP,更名為ChREBP-α),而在位于外顯子4的下一個起始位點開始,翻譯產生包含687個氨基酸的蛋白(ChREBP-β)。葡萄糖刺激ChREBP表達存在前饋機制,葡萄糖首先誘導ChREBP-α的轉錄,其反式激活ChREBP-β的轉錄,而后者則是更有效的轉錄激活因子。這種機制表明ChREBP-α不調節其自身的表達,而是有效地誘導另一種ChREBP同種型的表達。目前尚不清楚ChREBP-α的作用是否限于ChREBP-β,或兩種同工型是否與其靶基因的ChoRE相互作用并協同結合。
2 ChREBP表達調控和活性調節
2.1 ChREBP基因轉錄的表達調節 ChREBP的轉錄調節作用有賴于Max樣蛋白X(Max like protein X,Mlx)的存在。ChREBP必須與Mlx形成異二聚體后,才能與靶基因的ChoRE結合,發揮其轉錄調節作用[8]。因此,Mlx是ChREBP重要的功能伴侶。Iizuka等[9]研究發現,給予Mlx的顯性負突變體,體內能抑制脂肪合成過程酶的表達,從而改善糖尿病小鼠的糖脂代謝。
2.1.1 轉錄因子肝X受體(liver X receptors,LXR)核受體LXR對ChREBP的基因轉錄具有激活作用。LXR是脂質合成過程中的重要調節因子,能被固醇所激活,可與維甲酸X受體形成異二聚體,作用于靶基因啟動子的RXR/LXR DNA位點,從而調節靶基因的轉錄。Cha等[10]研究發現LXR激動劑能夠上調肝臟ChREBP及其靶基因LPK的表達,提示LXR可通過調節ChREBP來促進肝臟脂質合成,在轉錄水平直接調控ChREBP的表達。Mitro等[11]研究認為,葡萄糖可以激活LXR的表達,經過高糖處理的HepG細胞內ChREBP mRNA的表達水平可升高1倍,故葡萄糖是LXR的配體。但是Denechaud等[12]發現在LXRα/β聯合基因敲除小鼠肝臟,葡萄糖誘導的ChREBP、ACC、FAS基因表達與野生小鼠相比無差異,據此推斷,LXR并不是葡萄糖誘導肝臟脂質合成基因表達所必需。
2.1.2 多不飽和脂肪酸(polyunsaturated fatty acids,PUFA)PUFA對CHREBP的轉錄具有負調節作用。PUFA是肝內糖酵解和脂肪從頭合成過程中的抑制劑,抑制糖酵解及脂肪合成相關基因的表達如肝臟丙酮酸激酶(liver pyruvate kinase,LPK)和脂肪酸合成酶(fatty acid synthase,FAS)和乙酰輔酶A羧化酶(acetyl-CoA carboxylase,ACC),從而PFUA促進合成及儲存的脂肪酸氧化降解,是脂肪酸合成及降解的轉化調節劑。Dentin等[13]通過體內和體外實驗證實PFUA抑制CHREBP的活性,PFUA可加速CHREBP mRNA的降解,并且可改變CHREBP從胞質到核內的轉移。
2.1.3 甲狀腺激素、內毒素及細胞因子Hashimoto等[14]通過飲食改變和注射甲狀腺激素構建甲狀腺中毒小鼠模型,發現甲狀腺激素可通過TR-β1上調肝臟CHREBP mRNA及蛋白水平的表達。對ChREBP啟動子上可能存在的LXR結合位點進行研究發現,LXRE2的作用更為強大。在人類Hepal-6細胞和HepG2細胞中也證實了甲狀腺激素可以上調ChREBP mRNA和蛋白的表達水平,此過程由LXR調節,其調節方式與小鼠有很大差異。
Feingold等[15]發現在普通及高糖飲食情況下,內毒素、脂多糖(LPS)均可使肝臟ChREBP的表達下調;此外,酵母多糖和松節油也可下調肝臟ChREBP及其靶基因的表達;TNF-α和IL-1β能降低肝臟中ChREBP的表達。
Chau GC等[16]的最新研究則發現,缺乏哺乳動物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)的小鼠,表現出胰島β細胞團塊減少和胰島體積縮小。其機制是由于mTOR與ChREBP-Max樣蛋白復合物相關聯,并抑制了其轉錄活性。Chau等[16]也證明了mTOR可以通過與ChREBP-Mlx復合物相結合來調節凋亡機制以抑制TXNIP,減少β細胞凋亡及氧化應激,從而保護糖尿病環境中的胰腺β細胞。
2.2 ChREBP的活性調節
2.2.1 ChREBP的磷酸化對其活性的調節ChREBP活性的調節主要發生在DNA結合或轉錄活性的激活及從細胞質向細胞核的轉移兩個方面。ChREBP含有的磷酸化位點為Ser196、Ser626、Thr666和Ser568。Ser196位點主要調控ChREBP核質間轉移,而Ser626、Thr666和Ser568位點主要參與ChREBP結合DNA活性的調節[2]。蛋白激酶A(protein kinase A,PKA)對Ser196位點磷酸化作用可使ChREBP停留在細胞質中,而Ser196位點磷酸化的ChREBP經蛋白磷酸酶2A(protein phosphatase 2A,PP2A)去磷酸化后,進入到細胞核內。Thr666位點被PKA磷酸化后,ChREBP結合DNA的活性消失,而無活性的ChREBP經過PP2A去磷酸化后,結合DNA的活性又被激活。Ser568是AMP激活的蛋白激酶(AMP-activated protein kinase,AMPK)的特異性磷酸化位點,Ser568的磷酸化導致ChREBP結合DNA活性的喪失。而Ser626位點對調節ChREBP結合DNA的活性中僅起輔助作用。葡萄糖通過旁路代謝產物5-磷酸木酮糖促使細胞質中ChREBP的Ser196位點去磷酸化并進入細胞核內。ChREBP進入細胞核內后,葡萄糖通過核PP2A催化Thr666位點去磷酸化,激活無活性的ChREBP,在Ser196和Thr666去磷酸化后,活性的ChREBP與LPK基因ChoRE序列結合,激活LPK基因轉錄[17]。
2.2.2 葡萄糖傳感組件(glucose-sensing module,GSM)對其活性的調節 Tsatsos等[18]認為葡萄糖對ChREBP的調節并不依賴于PKA的磷酸化作用,而是通過另外的機制促進ChREBP表達。他們發現,低糖和高糖作用并不改變ChREBP的磷酸化水平,ChREBP磷酸化位點突變后,仍能對葡萄糖做出應答。Li等[19]認為葡萄糖對ChREBP的調控受GSM調節。GSM由低糖抑制區(LID)和葡萄糖反應活化保守元件(GRACE)2部分組成;低糖時,LID抑制GRACE的活性,高糖時,GRACE抑制LID的活性,認為GSM以這種方式調節ChREBP對葡萄糖的敏感性。ChREBP-α是全長ChREBP蛋白,而較短的同種型ChREBP-β則缺乏大部分LID,僅作為組成型活性蛋白,其表達與葡萄糖濃度無關[20]。
Dentin等[21]認為葡萄糖的代謝產物導致了ChREBP的活化,利用葡萄糖激酶(GK)敲除小鼠模型證實,GK的催化產物6-磷酸葡萄糖在ChREBP的活化及核內轉移中發揮重要作用。
2.2.3 ChREBP的乙酰化對其活性的調節Bricambert等[22]研究認為組蛋白乙酰轉移酶(histone acetyltransferase,HAT)的共激活物p300和鹽誘導基因 (Salt induced kinase2,SIK2)兩者共同作為ChREBP上游調節物以調節ChREBP的活性。ChREBP主要的乙酰化位點為Lys672,其位于ChREBP的DNA結合區域中,將Lys672位點突變后,ChREBP的DNA結合和轉錄激活活性都會有明顯的降低。可以推斷ChREBP的乙酰化對其作用于靶基因的啟動子是需要的。
2.2.4 肝受體同源物1(LRH-1)對其活性的調節近年來,Oosterveer MH的研究也提出,作為膽固醇代謝和膽汁酸穩態調節劑的LRH-1已經被認為是GK-ChREBP軸的上游調節因子,并作為響應于葡萄糖的肝代謝的新型調節劑[23]。這些發現表明ChREBP活性也可以通過LHR-1激活劑和/或抑制劑進行調節。
3 ChREBP的功能
3.1 ChREBP在肝臟中的功能 在肝臟中,存在許多參與碳水化合物和脂質代謝的關鍵酶,其轉錄水平會被高碳水化合物飲食等因素誘導。這些酶包括用于糖酵解的葡萄糖激酶(GK)和丙酮酸激酶(L-PK),ATP檸檬酸裂解酶,乙酰輔酶A羧化酶(ACC),脂肪酸合成酶(FAS)和用于脂肪生成的硬脂酰CoA去飽和酶(SCD1)以及用于戊糖磷酸途徑的葡萄糖-6-磷酸脫氫酶(G6PDH)(圖2)[20]。當體內攝入過多的碳水化合物時,在葡萄糖和胰島素的作用下,肝臟能通過從頭脂肪生成途徑將多余的碳水化合物轉化為三酰甘油,后者被運送至肝外的脂肪組織中貯存。在葡萄糖調節肝臟糖酵解與脂肪生成的過程中,ChREBP發揮了主要作用。
ChREBP是直接激活LPK的主要轉錄因子,通過激活LPK的表達,促進肝臟糖酵解。體外利用原代培養的小鼠肝細胞研究表明,過表達ChREBP能夠上調其LPK的表達水平;而利用RNA干擾的方法,下調ChREBP的表達則可阻斷高濃度葡萄糖誘導的LPK基因的表達上調[1]。染色質免疫共沉淀實驗證實,ChREBP能與LPK等靶基因的啟動子序列結合[24]。體內研究發現[2],在正常飲食條件下,ChREBP基因敲除小鼠的LPK表達水平下調、丙酮酸/磷酸烯醇式丙酮酸比率下降、丙酮酸的生成減少、糖酵解明顯受抑,同時肝臟的6-磷酸葡萄糖和糖原的含量增多;給予高糖飲食時,ChREBP基因敲除小鼠的肝臟質量約比對照小鼠重40%,推測可能與肝臟糖原的累積增多有關。這些研究結果證實,ChREBP是促使葡萄糖酵解并向脂質及糖原轉化的關鍵轉錄因子[25-26]。
ChREBP還可以激活ACC和FAS,從而促進肝臟的脂肪酸合成。ACC和FAS基因的啟動子區均含有ChoRE元件,介導了ChREBP的結合與轉錄激活作用。在普通飲食或高糖飲食情況下,ChREBP基因敲除小鼠肝臟中的脂質合成酶,包括ACC、FAS、ATP檸檬酸裂解酶(ATP citrate lyase,ACL)和脂酰CoA去飽和酶1(stearoyl-CoA desaturase-1,SCD1)等的mRNA表達水平均較對照小鼠明顯降低,導致其肝臟中由葡萄糖轉化而來的脂質成分降低大約65%,體內脂肪組織含量相對較低[2]。

圖2 ChREBP在控制肝細胞糖酵解及脂肪合成中的作用[21]
3.2 ChREBP在胰島組織中的功能 ChREBP廣泛表達于哺乳動物的各種組織中,但在脂肪合成器官,如肝臟、小腸和白色脂肪組織中有較高的表達。非常有趣,ChREBP在胰島中也有表達。在胰島中,葡萄糖不但刺激胰島素的分泌,而且還是許多細胞事件的重要信號。運用DNA微陣列的方法,許多研究者已經證實在胰島中葡萄糖應答的基因與在肝臟中的相似。在生成胰島素的INS-1細胞中過表達ChREBP能夠上調其LPK、FAS和ACC1的mRNA水平,但是細胞對于葡萄糖刺激后的胰島素水平與對照組相比沒有明顯變化。
Poungvarin等[27]研究發現,ChREBP作為一個轉錄因子,在高血糖誘導的β細胞糖毒性中起了關鍵性的作用。無論在體內或是體外,ChREBP均能激活其下游靶基因,包括FAS和硫氧還原蛋白結合蛋白(thioredoxin-interacting protein,TXNIP),其能導致脂質聚集,活性氧簇(ROS)在胰島細胞中過度產生,氧化應激增加,胰島素基因的轉錄減少和胰島細胞的凋亡。與非糖尿病患者相比,在糖尿病患者胰島的β細胞核中,ChREBP的免疫反應增加明顯。同時,由于糖尿病病程中長期存在的高血糖和高血脂,通常用作底物和代謝燃料的葡萄糖和游離脂肪酸(FAA)產生細胞毒性作用,從而導致胰島β細胞凋亡和腎衰竭,這種細胞功能的惡化被稱為葡萄糖-脂毒性,其致病機制可能為氧化應激和炎癥反應。胰腺外源性表達ChREBP的組成型活性形式時,在小鼠中觀察到葡萄糖不耐受和低效的胰島素分泌。
Chau等人[16]的最新研究也證明了哺乳動物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)可以通過與ChREBP-Mlx復合物相結合以調節凋亡,繼而抑制TXNIP的表達,從而保護糖尿病環境中的胰腺β細胞。
3.3 ChREBP在脂肪組織中的功能 ChREBP在脂肪組織中也有較高的表達。He等[28]研究表明,分化的3T3-L1脂肪細胞和大鼠脂肪組織表達ChREBP,胰島素、葡萄糖和抗糖尿病藥物troglitazone上調ChREBP在3T3-L1脂肪細胞的表達,而脂肪酸抑制其表達;禁食后再飼喂高碳水化合物膳食使大鼠脂肪組織ChREBP mRNA水平提高10倍。實驗發現在大鼠和豬前體脂肪細胞不表達ChREBP mRNA,分化中期開始表達;低糖條件下,胰島素對培養的成熟脂肪細胞ChREBP轉錄表達沒有影響,但在高糖培養基,胰島素明顯提高ChREBP和FAS及ACC1 mRNA表達水平;高濃度葡萄糖促進,而地塞米松抑制ChREBP轉錄表達;TNF-α明顯下調ChREBP轉錄表達,胰島素對此下調作用沒有明顯影響。對3T3-L1和原代培養脂肪細胞ChREBP表達的研究初步說明,與肝細胞類似,ChREBP也是脂肪細胞生物學作用的重要調節因子[29]。
Herman等[30]研究發現脂肪組織中的ChREBP在整合脂肪細胞和整個機體代謝功能中起到了很重要的作用,而這種作用是由ChREBP-β亞型的轉錄調節所介導。脂肪組織中的ChREBP對于葡萄糖的穩態具有有益的作用,其原因是由于其上調脂質的從頭合成(de novo lipogenesis,DNL)或是調節脂肪因子的功能。在人類研究中顯示,脂肪中的ChREBP表達與胰島素的敏感性具有直接而強有力的聯系,這也提示ChREBP在脂肪中的作用與調節機體的胰島素功能密切相關。
4 ChREBP與疾病的關系
4.1 ChREBP與代謝綜合征的關系 研究發現,脂肪肝與胰島素抵抗和糖尿病密切相關,是代謝綜合征的重要組成部分。鑒于ChREBP具有促進肝臟脂肪合成的作用,推測其與ob/ob小鼠的脂肪肝及胰島素抵抗密切相關。Dentin等[31]研究發現,在饑餓或飽食情況下,ob/ob小鼠肝臟ChREBP的表達水平均顯著升高,并且核內活性形式的ChREBP也顯著增加。在飽食情況下,ob/ob小鼠肝臟內ChREBP及SREBP-1c的表達都升高,從而引起脂肪合成增加,最終導致脂肪肝的形成。在饑餓情況下,ob/ob小鼠僅有ChREBP表達的上調,提示在饑餓的情況下,可能僅有ChREBP對ob/ob小鼠肝臟內脂肪合成發揮調控作用。
過多的脂質堆積在肝臟中會損傷肝細胞內的胰島素信號轉導通路,致使肝細胞對胰島素形成抵抗,而抑制肝細胞內的脂質堆積則可明顯改善肝細胞胰島素抵抗程度。敲除ob/ob小鼠體內的ChREBP基因后小鼠體質量明顯下降,同時胰島素抵抗、脂肪肝及葡萄糖耐受不良等表型都有明顯改善[32]。ob/ob小鼠肝細胞內異常表達的糖酵解和脂質合成基因在敲除了ChREBP后得以糾正,提示ChREBP對脂肪合成酶基因的誘導表達起到決定性的作用。將ChREBP基因敲除的小鼠雜交到ob/ob小鼠時,后者獲得了類似的保護性表型。這表明在肥胖或脂質堆積的情況下肝臟中ChREBP表達的改變有益于增加胰島素敏感性。Dentin等[31]研究表明,特異性抑制肝臟ChREBP可以糾正ob/ob小鼠脂肪肝和葡萄糖耐受力的下降,緩解小鼠的肥胖、胰島素抵抗和代謝綜合征癥狀。在感染ad-shChREBP的小鼠肝細胞內,萄萄糖-6-磷酸酶(glucose-6-phosphatase,G6Pase)活性的下降可降低肝臟的葡萄糖輸出,從而改善葡萄糖耐受力的降低[33]。總之,激活或抑制肝臟ChREBP對胰島素敏感性的影響機制復雜,因為它可能高度依賴于遺傳、飲食或環境因素。以上有關肝特異性敲除ChREBP動物模型的研究結果為進一步深入闡述明其在代謝綜合征中的關鍵作用提供了重要的實驗依據。另外,王冰等[34]學者近年研究顯示在1型糖尿病小鼠的腎臟中,FAS和ChREBP的表達量均較非糖尿病小鼠降低,胰島素可上調糖尿病小鼠腎臟FAS和ChREBP的表達水平。
4.2 ChREBP與腫瘤的關系 腫瘤細胞代謝模式的改變為細胞增生供能。大多數轉化細胞攝取高水平的葡萄糖通過有氧糖酵解產生ATP。在有氧糖酵解的過程中,糖分子中大部分碳原子參與脂質的重新合成和核苷酸合成。ChREBP作為葡萄糖反應轉錄因子在肝細胞內啟動新的葡萄糖代謝模式,在非增生性肝細胞的脂質合成中發揮重要的作用。Tong等[35]近年來研究發現,ChREBP能被絲裂原刺激誘導表達,對于細胞的高效率增生必不可少。抑制ChREBP的表達,會導致有氧糖酵解,脂肪酸從頭合成和核苷酸的生物合成都會減少;但可刺激線粒體呼吸,提示細胞從有氧糖酵解代謝轉變為氧化磷酸化。在體外實驗中chREBP受抑制的細胞內出現p53重新活化和細胞周期阻滯。而在體內抑制ChREBP可導致p53依賴的腫瘤生長減緩。這些研究結果表明chREBP在糖代謝轉向有氧模式和抑制p53活性中發揮重要的作用。
耿西林等學者[36]近年分別用qRT-PCR、免疫組化、Westernblot法檢測肝癌組織與癌旁組織以及多種HCC細胞系與正常肝細胞系中ChREBP的mRNA與蛋白表達發現:ChREBP的mRNA與蛋白表達在肝癌組織中表達均明顯高于癌旁組織,在所有HCC細胞系中均明顯高于正常肝細胞系。干擾ChREBP表達后,HCC細胞發生明顯G1-S期阻滯及細胞增生明顯降低,但細胞凋亡未發生明顯變化。即ChREBP在HCC中表達升高,可能通過調控細胞周期促進HCC細胞的增生,從而在HCC的發展中起了重要的作用。近年亦有證據表明ChREBP的表達和/或活性在腫瘤的早期和晚期階段有不同的調節。ChREBP在永生化造血細胞中有絲分裂時受到誘導表達,而在非小細胞肺癌的TGF-β1/Snail1介導的上皮-間質轉化(EMT)期間,其表達則有所抑制[35,37]。
4.3 ChREBP與Williams-Beuren綜合征 最初的研究發現遺傳性Williams-Beuren綜合征缺失17個基因,ChREBP是其中之一。該疾病主要表現為心臟、神經系統和皮膚的異常,約75%的患者表現出糖耐量降低和隱性糖尿病。Cairo等[4]研究發現ChREBP在細胞增生或分化中發揮重要作用。作為新發現的Mlx結合因子,可能導致Williams-Beuren綜合征的某種癥狀,但具體表現及機制目前尚不清楚。
5 展望
目前,脂肪肝、胰島素抵抗、肥胖等代謝性疾病嚴重危害著人類的健康,探討這些疾病的發生機制具有重要的意義。ChREBP的發現加深了對肝細胞中糖類和脂類代謝調節的了解,ChREBP-Mlx異二聚體調控肝葡萄糖利用和能量平衡,并且ChREBP在肝脂肪變性、肥胖癥、2型糖尿病、胰島素耐受性等方面起重要作用。ChREBP的發現已將葡萄糖作為信號分子與多種葡萄糖依賴性轉錄調控途徑相聯系,特別是一些參與糖酵解和脂肪生成過程的基因,其還參與了其他信號傳導和代謝途徑,并參與了腫瘤的發生。隨著分子生物學及人類基因組學的發展,人們對疾病的機制及信號通路機制有了更深入的認識,而分子靶向藥物治療也為越來越多的患者帶來福音。ChREBP有望成為肥胖等疾病治療的新靶點。進一步了解ChREBP對葡萄糖反應基因的調控機制有助于進一步研究代謝綜合征的病理機制,對治療相關疾病提供新的思路和手段[38]。通過加深對ChREBP的認識和了解,可能會有助于我們在未來找到某些代謝性疾病及惡性腫瘤治療的新機會。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
人人健康(2023年26期)2023-12-07 03:55:46
學苑創造·A版(2020年9期)2020-10-13 09:41:02
中國生殖健康(2019年2期)2019-08-23 08:12:10
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
中國衛生標準管理(2015年1期)2016-01-14 03:41:27
藥學與臨床研究(2015年4期)2015-06-05 11:35:51
中國醫藥科學(2015年15期)2015-02-27 12:32:27
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
中國全科醫學(2013年36期)2013-01-25 06:20:58
