Gaussian 09軟件在配合物紫外-可見吸收光譜教學中的應用
2018-10-18 07:16:10吳杰穎
赤峰學院學報·自然科學版 2018年9期
關鍵詞:實驗
張 瓊,吳杰穎
(安徽大學 化學化工學院,安徽 合肥 230601)
Gaussian軟件包用于執行各種半經驗和從頭分子軌道計算,是目前應用最廣泛的計算化學軟件之一[1],Gaussian 09軟件是Gaussian系列計算軟件的最新版本,其主要功能可用來預測分子在真空或者溶液狀態下的優化結構、電子排布、紫外-可見吸收光譜、紅外和拉曼光譜、核磁譜、分子極性等性質.Gaussian View是Gaussian軟件的圖形用戶界面,它主要的用途有兩個方面:(1)構建輸入文件;(2)直觀顯示計算結果.對于要用Gaussian軟件研究的體系,首先要在Gaussian View中構建分子結構,保存為.gjf文件,然后輸入Gaussian程序中計算,在構建的過程中,需要注意鍵長、鍵角、空間構型和對稱性等方面的準確性.此外,Gaussian View與多種圖形軟件兼容,可以讀入Chem3D和晶體數據等多種格式文件,大大拓寬了它的使用范圍.
金屬配合物的分子軌道是由金屬的價軌道和配體對稱性匹配的群軌道線性組合而成,涉及成鍵、反鍵、非鍵等多種軌道類型.最低激發態的電子構型受配位方式影響.圖1為過渡金屬配合物(八面體構型)的分子軌道能級示意圖[2],金屬到配體的電荷轉移(MLCT)是電荷從金屬d軌道躍遷到配體π*軌道,配體到金屬的電荷轉移(LMCT)是電荷從配體轉移到金屬.其他躍遷方式包括配體到配體電荷躍遷(LLCT)、配體內電荷躍遷(ILCT)及金屬中心(MC)電荷躍遷.
電子的躍遷在紫外-可見吸收光譜的教學過程中較抽象,很難靠傳統的教學方法獲得良好的教學效果.將Gaussian軟件應用于配合物的紫外-可見吸收光譜教學中,具體做法是:以課題組合成的多種過渡金屬配合物為研究對象,利用配合物的紫外-可見吸收光譜的實驗結果[3-4],在單晶結構基礎上,采用含時密度泛函理論(TD-DFT)計算配合物的躍遷.結構優化采用未進行對稱限制的B3LYP泛函進行,然后在優化結構的基礎上采用B3LYP泛函進行TD-DFT計算.結構優化及TD-DFT計算均采用Gaussian 09程序進行.對于基態的優化及最低的25個單線態到單線態的激發能量的TD-DFT計算采用6-31G*基組 (對于C、H、N、O 原子)及 LanL2dz 基組(對于 Pt、Zn、Cl、Br原子).理論結合實驗,可以幫助學生更形象地理解金屬配合物的電子躍遷方式,收到較好的教學效果.

圖1 八面體構型配合物的分子軌道能級示意圖
1 配體內的電荷躍遷

圖2 (a)TDPt結構 (b)由實驗獲得配體TSD及配合物TDPt的紫外-可見吸收光譜(c-d) 通過Gaussian計算獲得TDPt的前線分子軌道電子云分布圖
配體的π軌道中的電子受激發躍遷到反饋π*空軌道(π→π*)稱為配體內的電荷躍遷(intraligand charge transfer,ILCT),ILCT對應的紫外-可見吸收特征峰一般在紫外區.ILCT具有強吸收,ε值在104數量級,金屬中心對ILCT干擾小,紫外-可見吸收特征峰幾乎不受其他輔助配體影響.
紫外-可見吸收光譜結果表明:配合物在310nm處的紫外可見吸收光譜ε值為5×104mol-1L cm-1,僅比配體在305nm處的吸收峰紅移5nm,可歸屬于ILCT.理論計算結果表明:在配合物分子HOMO-1軌道中,電子云主要分布在三苯胺官能團,在LUMO+2軌道中,電子云分布在N,N二乙基苯胺官能團,電子云從HOMO-1軌道躍遷至LUMO+2軌道,可明顯觀察到ILCT,通過軌道間的能級差ΔE計算出的紫外-可見吸收光譜特征峰的位置與實驗結果吻合,為解釋電子躍遷提供了有力的證明.
2 金屬到配體的電荷躍遷
金屬中心d軌道到有機配體π*軌道的躍遷稱為金屬到配體的電荷躍遷 (metal-to-ligand charge transfer MLCT),金屬中心d軌道和配體π*軌道較低能級間隙導致紫外-可見吸收特征峰出現在長波區.MLCT通常包含容易氧化的金屬中心和具有低能空軌道的配體.MLCT具強吸收,ε值在104數量級或更大,金屬中心和配體會影響MLCT特征峰,調節配體可調控峰的位置.
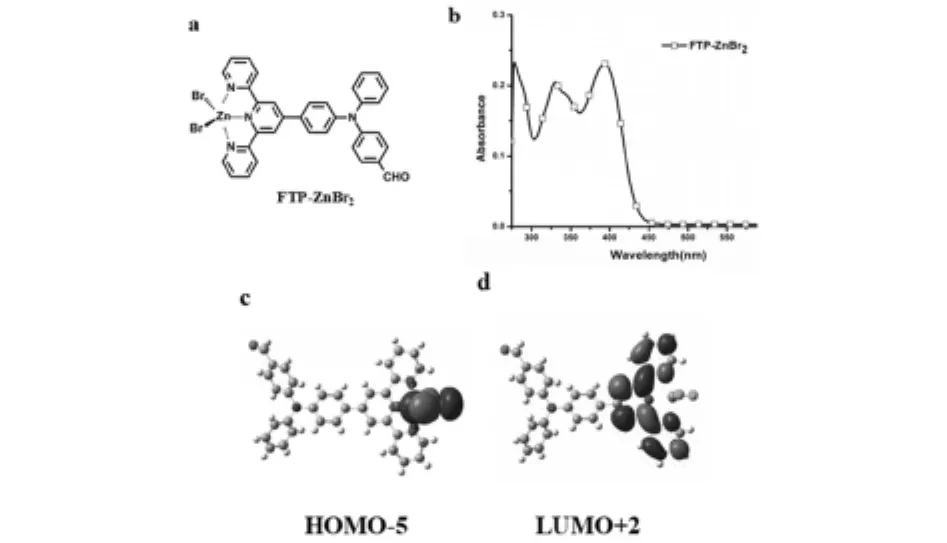
圖3 (a)FTP-ZnBr2結構 (b)由實驗獲得FTP-ZnBr2的紫外-可見吸收光譜 (c-d)通過Gaussian計算獲得FTP-ZnBr2的前線分子軌道電子云分布圖
紫外-可見吸收光譜結果表明:配合物在405nm處的紫外-可見吸收光譜ε值為2.5×104mol-1L cm-1,可歸屬于MLCT.理論計算結果表明:在配合物分子的HOMO-5軌道中,電子云主要分布在中心金屬原子(Zn),在LUMO+2軌道中,電子云分布在三吡啶官能團,可以明顯地觀察到MLCT,通過HOMO-5與LUMO+2的能級差ΔE計算出的紫外-可見吸收光譜特征峰為410nm,與實驗值基本一致.
3 配體到配體的電荷躍遷
當配合物中含有兩種或多種配體時,其中一種配體最高占有軌道能級高于金屬離子的最高占有d軌道,產生配體到配體的電荷躍遷 (Ligand-to-ligand charge transfer LLCT).LLCT的吸收一般屬中等強度吸收,ε值一般在103~104數量級.LLCT躍遷受配體的取代基影響較大,不同的供吸電子基團可使紫外-可見吸收特征峰發生紅移/藍移.
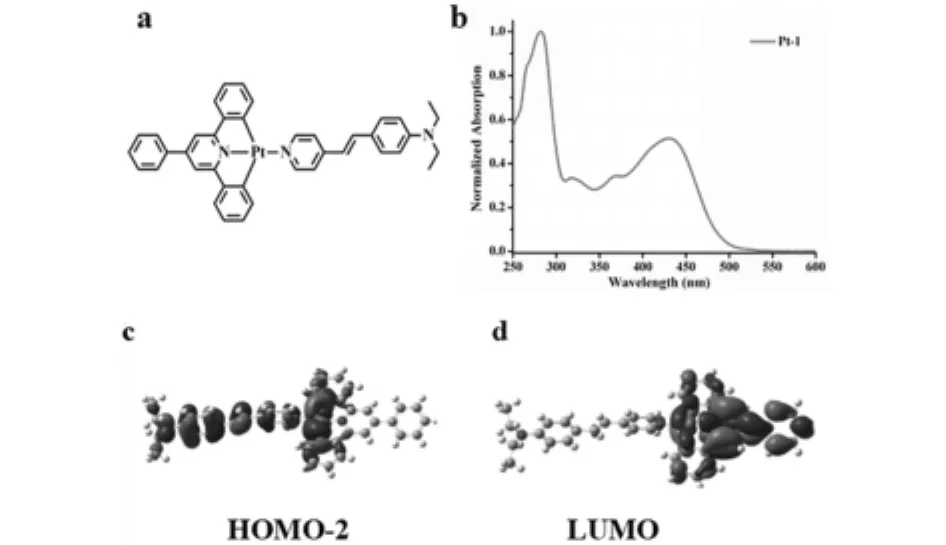
圖4 (a)Pt-1結構 (b)由實驗獲得Pt-1的紫外-可見吸收光譜 (c-d)通過Gaussian計算獲得Pt-1的前線分子軌道電子云分布圖
紫外-可見吸收光譜表明:Pt-1在320nm和375nm處的吸收峰的ε值大約為4×104mol-1L cm-1,可以歸屬為LLCT.理論計算結果表明:在Pt-1的HOMO-2軌道中,分子的電子云主要分布在N,N-二乙基苯乙烯基吡啶上.在LUMO軌道中,分子的電子云主要分布在6'-苯基-2,2'-聯吡啶上,電子從HOMO-2軌道躍遷至LUMO軌道,即LLCT,通過軌道間能級差ΔE計算紫外-可見特征峰位置與實驗結果相吻合.
綜上所述,在配合物的紫外-可見吸收光譜教學中,通過Gaussian軟件的引入能夠在教學過程中對于配合物中的電子激發態給學生提供更為形象的描述,通過理論計算,可以讓學生掌握電子云分布的抽象概念,也可以讓學生驗證實驗結果,極大地激發學生學習興趣,提高教學質量和教學效果.
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
