基于E6/E7基因檢測高危型人乳頭瘤病毒感染新方法的構建*
2018-11-08 07:24:50謝旺凱呂詩卉薛向陽陳向敏
檢驗醫學與臨床 2018年21期
關鍵詞:檢測
王 儉,謝旺凱,李 婷,呂詩卉,倪 超,薛向陽,陳向敏,陳 俊△
(溫州醫科大學:1.第一臨床醫學院;2.第二臨床醫學院;3.基礎醫學院;4.檢驗醫學院/生命科學院,浙江溫州 325035)
宮頸癌是一種嚴重危害女性健康的惡性腫瘤,位于女性惡性腫瘤的第二位[1]。研究已證實,幾乎所有宮頸癌都是由人乳頭瘤病毒(HPV)尤其是高危型 HPV感染引發,因此高危型HPV檢測逐漸成為宮頸癌篩查的研究熱點[2]。目前,HPV研究主要針對HPV基因組進行。HPV基因組可分為早期區、晚期區及上游調節區(URR)或長控制區(LCR)或非編碼區(NCR)。早期區編碼E6、E7、E1(E8)、E2 、E4 (E3)、E5等5~8個基因,晚期區編碼L1和L2病毒結構蛋白[3]。目前國內外廣泛采用HPV通用引物定位在HPVL1基因上進行檢測[4]。但是在高危型HPV感染過程中,其基因會整合到宿主細胞基因組中,這是引發宮頸細胞癌變發生的前提[5]。而高危型HPV DNA的整合過程往往會發生基因斷裂缺失,如E1/E2調控區和L1/L2衣殼蛋白基因的缺失,因此基于L1基因的檢測存在一定的漏診現象。然而,研究表明E6和E7基因在整合過程中卻可以保留完整,不易發生缺失,因此,E6/E7基因被認為是檢測HPV最為理想的靶點[6]。基于以上現象,本研究針對HPV E6/E7基因,設計我國感染率較高的7種高危型HPV特異性引物,包括HPV16、18、31、33、51、52、58,建立一種成本低廉、效率更高的檢測宮頸腫瘤、脫落細胞等組織高危型HPV感染的多重聚合酶鏈反應(PCR)檢測方法,并與現在臨床上主流的流式熒光雜交法進行比較,評價其臨床意義。
1 材料與方法
1.1標本來源
1.1.1宮頸腫瘤組織標本 68份宮頸腫瘤組織標本采自溫州醫科大學附屬第一醫院婦產科68例手術患者,并經病理檢查確診,其中宮頸上皮內瘤變(CIN) 1級12例,CIN 2級19例,CIN 3級19例,宮頸鱗癌12例,宮頸腺癌2例,正常宮頸(內膜透明細胞癌)4例,標本離體后保存于液氮中。患者年齡25~69歲,平均(42.4±1.1)歲。所有標本收集均征得患者及或家屬的同意,并簽署知情同意書。
1.1.2宮頸脫落細胞標本 150份宮頸脫落細胞標本均取自溫州醫科大學附屬醫院婦科門診患者。患者年齡18~78歲,平均(43.1±11.6)歲。所有標本收集均征得患者及或家屬的同意,并簽署知情同意書。HPV16陽性的Siha細胞和HPV18陽性的Hela細胞,購自美國ATCC,并由實驗室保存。
1.2DNA提取 宮頸腫瘤組織標本每份切取50 g病變區組織,碾碎后加入50 μL磷酸鹽緩沖液(PBS)中制成懸液備用;Siha、Hela細胞貼壁培養至長滿50 mL培養瓶后,也制成懸液備用。制備的懸液采用DNA提取試劑盒[TIANGEN TIANamp Genomic DNA Kit(DP304)]提取DNA。將DNA模板統一稀釋至100 ng/μL。宮頸脫落細胞溶于細胞保存液中,取2 mL細胞溶液10 000 r/min離心1 min后去上清液,采用DNA提取試劑盒提取DNA。
1.3引物
1.3.1引物設計 在分析同源性的基礎上,針對我國流行的主要高危型HPV16、18、31、33、51、52、58及相應HPV基因組的E6/E7基因,設計這7種HPV型別的引物。由北京六合華大基因科技有限公司合成[7]。引物序列見表1。將合成的引物稀釋成10 μmol/L,-20 ℃保存,待用。
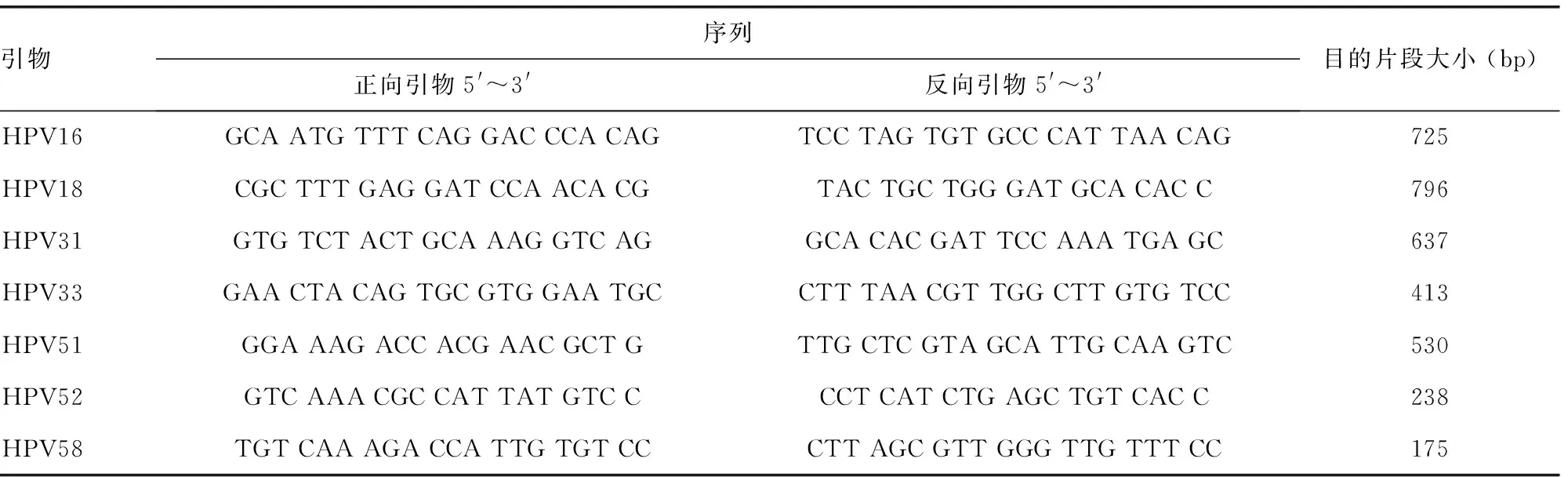
表1 各型別HPV特異性引物序列
1.3.2引物特異性驗證 為驗證相應HPV型別引物特異性,以明確HPV分型的宮頸腫瘤組織標本和細胞株的DNA為模板,引物FP 10 μmol/L,引物RP 10 μmol/L,Taq Mix 12.5 μL[TIANGEN 2×Taq PCR MasterMix(KT201)]配制成25 μL反應體系。退火溫度為63 ℃(16、18、31、33、51、58型),65 ℃(52型)。熱循環條件設置如下:94 ℃預變性5 min;94 ℃變性30 s;退火30 s;72 ℃延伸1 min,35個循環;最后72 ℃后延伸5 min。在Bio-Rad S1000 PCR儀上進行反應。PCR產物瓊脂糖凝膠電泳,切膠回收,基因測序,并與已公布的各型別HPV基因序列比較。
1.3.3引物敏感性驗證 為分析特異性引物敏感性, 以10.00、1.00、0.10、0.01 ng不同HPV型別陽性的DNA作為模板進行試驗,PCR產物進行瓊脂糖凝膠電泳,并進行敏感性分析。
1.4E6/E7多重PCR檢測方法的構建 以7種明確HPV分型的宮頸腫瘤組織標本為DNA模板,將HPV16、18、31、33、51、52、58型的特異性引物按濃度1∶1∶1∶1∶1∶1∶1的比例混合加入4 μL,RNase-free去離子水1 μL,Taq Mix 7.5 μL;Q-Solution+1.5 μL[QIAGEN Multiplex PCR kit(Cat.No.206143)]配制成15 μL體系。熱循環條件設置如下:95 ℃預變性15 min;94 ℃變性30 s;63 ℃退火90 s;72 ℃延伸45 s,35個循環;最后68 ℃后延伸15 min。PCR產物進行瓊脂糖凝膠電泳,對比各型別HPV的單一PCR產物電泳圖譜以確定多重PCR方法的特異性。在此基礎上,以10.00、1.00、0.10、0.01 ng不同HPV型別陽性的DNA作為模板進行試驗,PCR產物進行瓊脂糖凝膠電泳,并進行敏感性分析。同時,為避免實驗存在的偶然性,相隔一定量的時間后,同樣以新建立方法時所使用的7種高危型別陽性標本DNA作為模板,在相同條件下以同樣的參數進行實驗,驗證多重PCR方法的穩定性。
1.5宮頸腫瘤組織標本HPV感染型別檢測 對68份宮頸腫瘤組織標本采用所構建的多重PCR方法進行檢測。同時對相同標本采用流式熒光雜交法進行平行檢測,記錄并比較分析結果。
1.6宮頸脫落細胞標本檢測 使用新建立的多重PCR方法對150份宮頸脫落細胞標本進行HPV感染情況檢測,同時對相同的標本采用流式熒光雜交法進行平行檢測,記錄并比較分析結果。
1.7統計學處理 采用SPSS 21.0統計軟件進行數據處理及統計分析,計數資料以例數或率表示,組間比較采用χ2檢驗,P<0.05為差異有統計學意義。
2 結 果
2.1引物設計結果 引物特異性實驗表明7種高危型HPVE6/E7引物(HPV16、18、31、33、51、52、58)均具有特異性,見圖1。同時敏感性分析實驗顯示:HPV16、18、33型DNA 模板量低至1 ng仍能擴增特異目的片段,HPV31型DNA模板量低至10 ng仍能擴增特異目的片段,HPV51型DNA模板量低至0.1 ng仍能擴增特異目的片段,HPV52、58型DNA模板量低至0.01 ng仍能擴增特異目的片段。
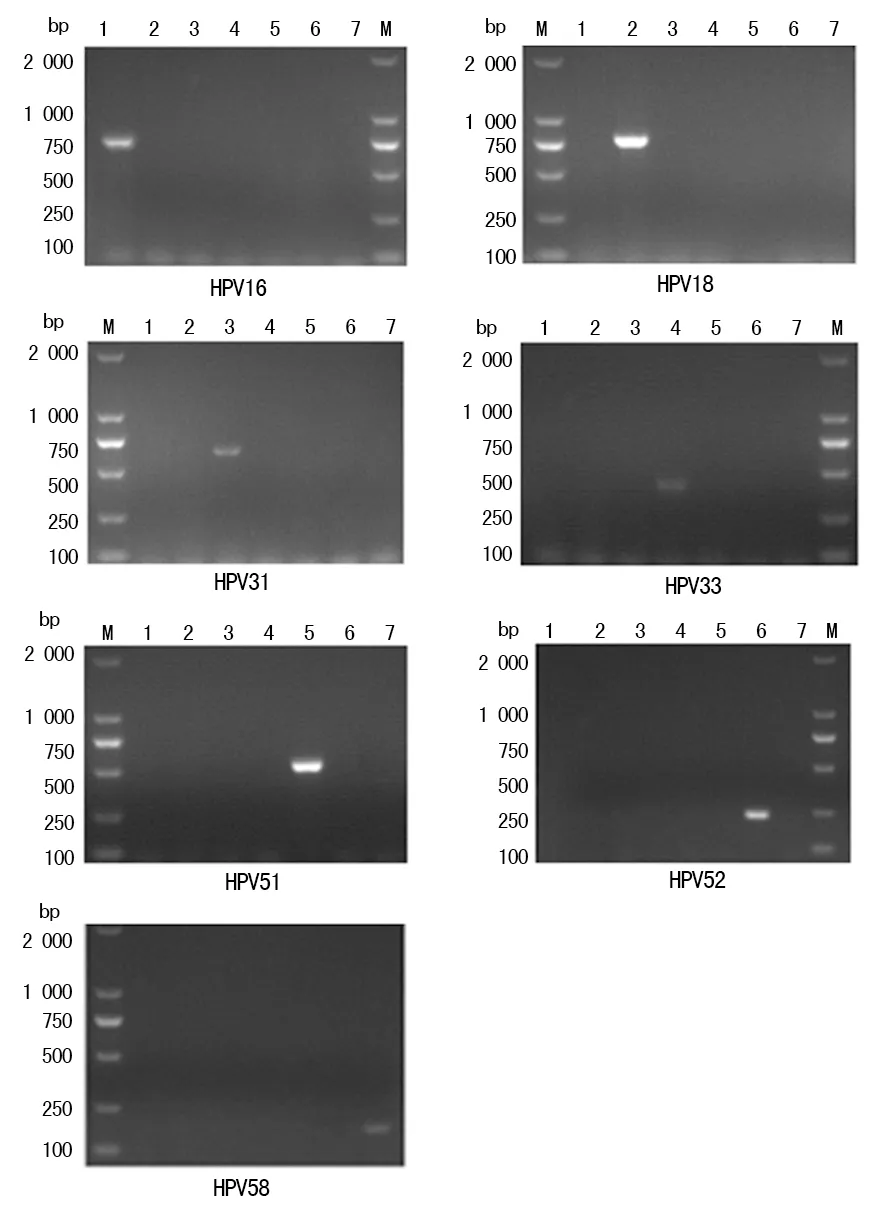
2.2E6/E7多重PCR檢測方法的建立 對已經明確了HPV分型的宮頸腫瘤標本和細胞株所提取的DNA進行多重PCR擴增,結果顯示出現單一條帶,與使用單一型別引物進行PCR的結果一致,提示所建立的多重PCR可特異檢測這7種HPV亞型,見圖2。且敏感性分析實驗顯示:HPV16、18、31、33、58型DNA模板量低至10 ng仍能擴增特異目的片段,HPV51和52型DNA模板量低至0.1 ng仍能擴增特異目的片段。
2.3宮頸腫瘤組織HPV檢測結果 以新建立的多重PCR方法檢測68份宮頸腫瘤組織標本,見圖3,根據電泳條帶判斷68份宮頸腫瘤組織標本HPV感染情況,檢測出HPV陽性標本48份(70.59%),其中HPV16陽性36份(52.94%),HPV18陽性3份(4.41%),HPV31陽性4份(5.88%),HPV33陽性7份(10.29%),HPV51陽性2份(2.94%),HPV58陽性3份(4.41%),多重感染6份(8.82%)。高于流式熒光雜交法檢測所顯示39.71%的感染率,2種方法檢測結果比較,差異有統計學意義(χ2=11.43,P<0.05)。
注:M為D2000 DNA標記物;1為HPV16陽性標本;2為HPV18陽性標本;3為HPV31陽性標本;4為HPV33陽性標本;5為 HPV51陽性標本;6為HPV52陽性標本;7為 HPV58陽性標本
圖1所設計的7種高危型別HPV E6/E7引物的特異性分析
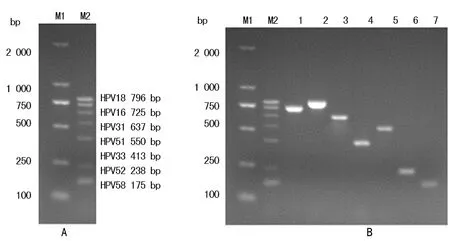
注:A為HPV陽性標準條帶;B為HPV 7種亞型引物特異性分析;M1為D2000 DNA 標記物;M2為陽性標準條帶;1為HPV16陽性標本;2為HPV18陽性標本;3為 HPV31陽性標本;4為HPV33陽性標本;5為HPV51陽性標本;6為HPV52陽性標本;7為HPV58陽性標本
圖2多重PCR法電泳結果分析
2.4宮頸脫落細胞標本檢測結果 150份宮頸脫落細胞標本用新建立的多重PCR方法及流式熒光雜交法檢測的結果顯示7種高危型別(HPV16、18、31、33、51、52、58)檢出率比較,差異無統計學意義(χ2=3.68,P>0.05),見表2。
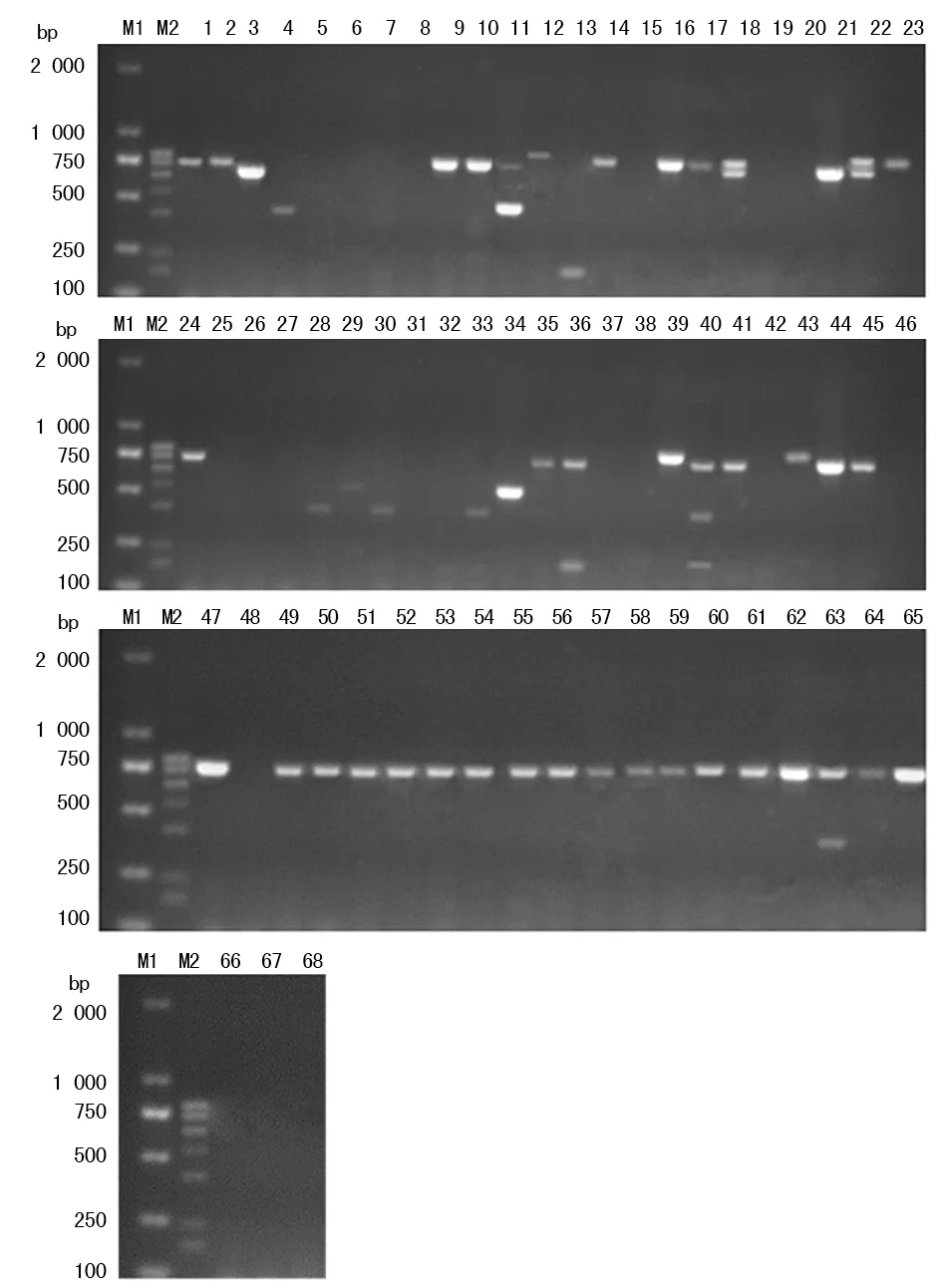
注:M1為 D2000 DNA標記物;M2為陽性標準條帶;1~68為宮頸腫瘤組織標本
圖3 68份宮頸腫瘤組織多重PCR檢測結果分析
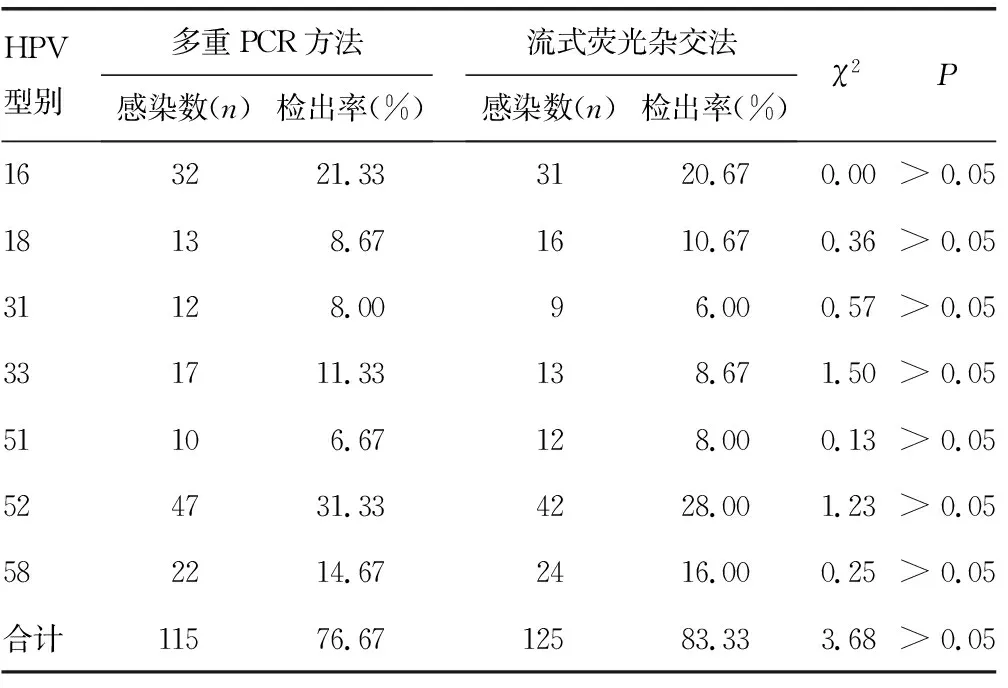
表2 150份宮頸脫落細胞不同檢測方法的HPV檢測結果(n=150)
3 討 論
高危型HPV持續感染是導致宮頸癌發生的重要原因。越來越多的研究表明HPV基因組檢測對宮頸病變預測和術后療效判斷有利,可以避免不必要的診斷和治療。因此,國內外采用了很多種方法對HPV感染進行檢測,應用于臨床宮頸癌篩查和防治。
現在臨床上采用較多的是基于L1衣殼蛋白基因的HPV檢測方法,如流式熒光雜交法[8]。但由于在HPV感染過程中,病毒 DNA通常會與宿主染色體整合,會改變整合位點及鄰近宿主基因的表達,使部分HPV L1基因組發生斷裂丟失,最終導致HPV L1衣殼蛋白也表達缺失,所以基于L1基因序列的HPV檢測方法容易出現假陰性的結果,這也限制了其在臨床上的使用[9]。而早期蛋白基因E6/E7在腫瘤的發生過程中持續存在,且其表達的E6/E7早期蛋白在宮頸癌變過程中起著重要作用。惡性腫瘤病變時,HPV DNA整合到宿主DNA當中,通常導致E2基因中斷從而使E6/E7基因表達增強[10]。E6/E7蛋白分別抑制P53、Rb基因的活性,激活人細胞端粒酶的轉錄,引起細胞異常分化,導致正常細胞永生化,最終形成腫瘤[11]。因此,對HPV E6/E7基因進行檢測,能從根源上對宮頸病變進行診斷和預防,是判斷HPV感染的更好選擇。然而,臨床上針對E6/E7的檢測方法不多,尤其是多重PCR的檢測方法幾乎沒有。為此本研究主要構建了基于E6/E7的多重PCR方法檢測HPV的感染情況,在對HPV進行同源性分析后證實各HPV亞型的E6/E7基因具有特異的保守片段,據此選擇設計并驗證了中國人群感染率較高的7種高危型別HPV E6/E7的特異性引物,優化條件后建立了一種基于E6/E7基因的HPV多重PCR分型檢測方法。對已明確型別的宮頸癌組織標本及宮頸細胞的檢測陽性標本出現條帶,而陰性標本未出現條帶證明其特異性好,而且實驗還驗證了該多重PCR技術檢測靈敏度能達到10.00 ng,技術穩定性良好,滿足臨床標本檢測方法的要求。
68份宮頸腫瘤組織標本的HPV檢測結果顯示,所構建的基于E6/E7多重PCR檢測陽性率明顯高于基于L1基因的流式熒光雜交法檢測,差異有統計學意義(P<0.05)。而對門診采集的150份宮頸脫落細胞標本檢測結果顯示7種高危型別檢出率與流式熒光雜交法檢出率并無差異,考慮到門診患者高危型別感染較少,樣本數不夠大,有待進一步驗證。
臨床上使用的基于L1基因的流式熒光雜交法檢測費用較高,操作復雜,需要大型儀器設備支持,不利于在鄉鎮衛生院開展。而本研究所建立的基于E6/E7多重PCR法不僅能夠一次檢測HPV16、18、31、33、51、52和58 7種高危型別,而且具有操作簡便、成本低廉和所需儀器較少且較易獲得等優勢,同時研究結果也已表明兩種方法檢出率高度相近。因此,基于E6/E7多重PCR法在應用中能極大彌補流式熒光雜交法在成本和技術等方面的不足,同時可以減少多次檢測帶來的交叉污染及高成本的問題,適合在臨床大面積開展患者高危型HPV感染的早期篩查。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
