p PB修飾的肉蓯蓉苯乙醇總苷脂質體的制備研究
2019-01-10 07:00:04木尼熱庫爾班由淑萍新疆醫科大學公共衛生學院新疆烏魯木齊830011
中國藥物應用與監測 2018年6期
木尼熱·庫爾班,王 梅,由淑萍,劉 濤(新疆醫科大學公共衛生學院,新疆 烏魯木齊 830011)
肉蓯蓉(Herba Cistanches)為列當科植物管花肉蓯蓉的干燥帶鱗葉的肉質莖,具有補腎、益經血、潤腸通便等作用,有“沙漠之參”之稱[1-3]。苯乙醇總苷是肉蓯蓉的主要化學成分之一,主要包含松果菊苷與毛蕊花糖苷兩種成分。研究顯示苯乙醇總苷能夠明顯抑制機體內酪氨酸酶的活性,并在修復自由基損傷、抗衰老、抗疲勞、抗色素沉著、抗老年癡呆癥方面顯示了強大的功效[3-5]。
肝纖維化(hepatic fibrosis,HF)是肝臟遭受反復或慢性損傷后的一種損傷與修復過程,主要與慢性炎癥和引起慢性肝臟損傷的化學因素相關。研究表明,肉蓯蓉苯乙醇總苷可降低肝纖維化大鼠肝組織中的膠原纖維生成,因此具有良好的抗肝纖維化的作用[6-8]。環肽pPB能特異性識別血小板源生長因子受體,因此能靶向到肝纖維化細胞。脂質體(liposomes)是由磷脂構成的雙分子層結構的微型囊泡,脂質體作為藥物載體具有緩釋、降低藥物毒性、保護被包封的藥物和靶向性等特點,以脂質體給藥后,其可將藥物自動轉運到肝、脾、肺和骨髓等組織器官,對于治療肝、脾部位疾病及提高藥物的治療指數具有很好的作用[9-11]。在上述基礎上,本研究擬將肉蓯蓉苯乙醇總苷制成pPB環肽修飾的脂質體,利用pPB環肽作用,使脂質體聚集到肝纖維化的細胞,更好地發揮肉蓯蓉苯乙醇總苷治療肝纖維化的作用。本研究采用不同的制備方法分別制備p PB修飾的肉蓯蓉苯乙醇總苷脂質體,并選擇松果菊苷為含量測定指標,對脂質體的粒徑、電位和包封率進行測定,最終確定該脂質體的制備方法[12-13]。
1 儀器與試藥
1.1 儀器
AB135-S分析天平(瑞士Mettler Toledo公司);T6新世紀紫外可見分光光度儀(北京普析通用儀器有限公司);EYELAN-2100旋轉蒸發儀(上海愛朗儀器有限公司);Malvern Nano-2S90型激光粒徑測定儀(英國馬爾文儀器有限公司);JEOL-1340H透射電子顯微鏡(捷歐路科貿有限公司);85-2型控溫磁力攪拌機(江蘇金怡儀器科技有限公司);KQ-500DE超聲波清洗器(昆山市超聲儀器有限公司)。
1.2 試藥
松果菊苷對照品(批號327A021,含量98%,北京索萊寶科技有限公司);苯乙醇總苷原料藥(實驗室自制,純度95%);大豆卵磷脂(Lipoid公司);二棕櫚磷脂酰膽堿(DPPC,意大利Corden Pharma公司,批號T0343);膽固醇(美國Sigma公司);馬來酰基-二硬脂酸磷脂酰乙醇胺-聚乙二醇2000(Mal-DSPE-PEG2000,美國Nanocs公司,批號130916);p PB(Cys*-Ser-Arg-Asn-Leu-lle-Asp-Cys*,批號20170204,含量98.49%,上海吉爾生化有限公司);N-琥珀酰亞胺基-S-乙酰硫基乙酸酯(SATA,美國Sigma公司,批號SLBR3300V);Sephadex G-50(100-300)(美國Sigma公司,批號SLBB6637V);其余試劑均為分析純。
2 方法與結果
2.1 含量測定方法的建立
2.1.1 最大吸收波長的選擇取適量甲醇溶解松果菊苷對照品,以甲醇作為空白,在紫外分光光度儀上200 ~ 400 nm范圍內進行紫外掃描,考察最大吸收波長,結果顯示在330 nm處松果菊苷有最大吸收。
2.1.2 標準曲線建立精密稱取松果菊苷對照品適量,用甲醇溶解并定容至刻度,得200 μg·mL-1的松果菊苷對照品儲備液。分別精密量取對照品儲備液適量,用甲醇溶解,制得濃度分別為5.0、8.0、10.0、20.0、40.0 μg·mL-1對照品溶液,將各溶液置于330 nm處測定吸光度值,以濃度(C)對吸光度(A)值作線性回歸,得線性回歸方程C = 42.615 A + 1.210 8,r =0.999 8,結果表明,在5.0 ~ 40.0 μg·mL-1范圍內線性關系良好。
2.1.3 精密度實驗配制10.0、20.0、40.0 μg·mL-1的低、中、高濃度的松果菊苷對照品溶液,每個濃度配3份,1天內重復測定3次,計算日內精密度,連續測定3 d,計算日間精密度。結果顯示日內精密度和日間精密度的RSD值均小于2%,表明方法精密度良好。
2.1.4 回收率考察取0.8 mL肉蓯蓉苯乙醇總苷脂質體作為樣品的加入量,并加入高、中、低濃度松果菊苷對照品溶液分別為0.5、1.0、2.0 mL,使對照品量分別為樣品加入量的120%、100%、80%,樣品與對照品溶液混合均勻后,用甲醇定容至刻度,用紫外分光光度計測定藥物含量,計算對照品的加入量及回收率。回收率(%)=(測得總量-樣品加入量)/實際對照品的加入量×100%。結果顯示松果菊苷的平均回收率分別為98.85%、100.67%、99.62%,RSD均小于2%,回收率符合規定,結果見表1。
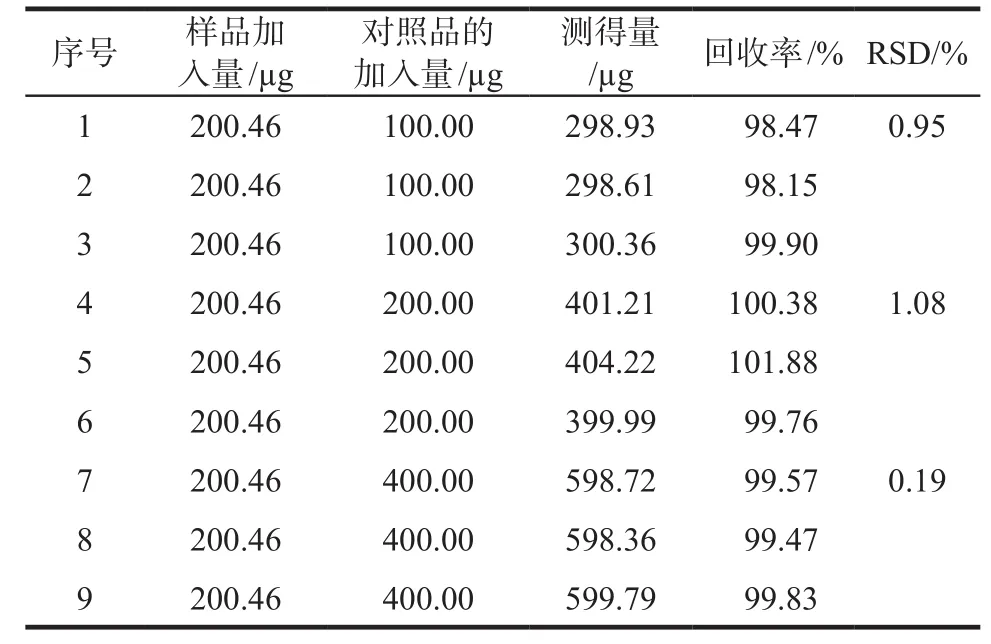
表1 苯乙醇總苷含量加樣回收率實驗結果Tab 1 Results of recovery rates of phenylethanoid glycosides
2.2 肉蓯蓉苯乙醇總苷脂質體的制備
2.2.1 薄膜分散法制備肉蓯蓉苯乙醇總苷脂質體稱取卵磷脂、DPPC、膽固醇適量(質量比為2∶2∶1),溶于適量氯仿和甲醇混合溶劑中(氯仿∶甲醇 = 4∶2,v∶v),置于旋轉蒸發儀,在49 ~ 50 ℃下旋轉蒸發使成膜狀,然后加入250 μg·mL-1的苯乙醇總苷水溶液旋轉水化,即得脂質體,將所得脂質體分別用0.45 μm微孔濾膜擠壓4 ~ 5次,0.22 μm的微孔濾膜擠壓18 ~20次,即得肉蓯蓉苯乙醇總苷脂質體。
2.2.2 二次包封法制備肉蓯蓉苯乙醇總苷脂質體稱取卵磷脂、DPPC、膽固醇適量(質量比為2∶2∶1),溶于適量氯仿和甲醇混合溶劑中(氯仿∶甲醇 = 4∶2,v∶v),置于旋轉蒸發儀,在49 ~ 50 ℃下旋轉蒸發使成膜狀,然后加入250 μg·mL-1的苯乙醇總苷水溶液旋轉水化,得脂質體備用。同法再次取磷脂成分溶于氯仿,旋轉成膜狀后,將制得備用的脂質體加入,再次旋轉水化,即得二次包封的脂質體。將所得脂質體分別用0.45 μm的微孔濾膜擠壓4 ~ 5次,0.22 μm的微孔濾膜擠壓18 ~ 20次,即得肉蓯蓉苯乙醇總苷脂質體。
2.2.3 逆相蒸發法制備肉蓯蓉苯乙醇總苷脂質體稱取質量比為2∶2∶1的卵磷脂、DPPC、膽固醇,溶于適量氯仿和甲醇混合溶劑中(氯仿∶甲醇 = 4∶2,v∶v),加入250 μg·mL-1苯乙醇總苷原料藥水溶液適量,冰浴超聲1 h,直至形成穩定的乳劑,采用旋轉蒸發儀減壓蒸發,達到膠態后,加適量水水化,繼續減壓蒸發除去殘留溶劑,將所得脂質體分別用0.45 μm的微孔濾膜擠壓4 ~ 5次,0.22 μm的微孔濾膜擠壓18 ~20次,即得肉蓯蓉苯乙醇總苷脂質體。
2.2.4 pPB-DSPE-PEG2000的制備首先將環肽pPB分子中引入巰基(-SH),然后使p PB環肽上巰基與Mal-DSPE-PEG2000末端的馬來酰基發生C-S加成反應。具體方法為:將4 mg pPB溶于PBS緩沖液,并與8 mg SATA混合,使SATA與pPB質量比為2 : 1,在室溫下攪拌2 h后,加入Mal-DSPE-PEG2000,室溫下磁力攪拌24 h,將所得反應產物用截留分子量為3500Da的透析袋透析除去多肽、SATA等,截留下鏈接好的產物p PB-DSPE-PEG2000,將產物冷凍干燥后備用。為檢測偶聯是否成功,將p PB-PEG2000-DSPE和Mal-PEG2000-DSPE分別溶于氚代氯仿中,分別測定1H-NMR,通過比較馬來酰基特征峰(6.7 ppm)的消失與否來驗證偶聯是否成功。結果表明pPB-PEG2000-DSPE1H-NMR圖譜中6.7 ppm未見相應的馬來酰基峰出現,說明偶聯已成功。
2.2.5 pPB修飾的肉蓯蓉苯乙醇總苷脂質體的制備稱取大豆磷脂、DPPC、膽固醇、pPB-DSPE-PEG2000(質量比為10 : 10 : 5 : 1)溶于氯仿和甲醇的混合溶劑,同“2.2.2”項下方法制備pPB修飾的肉蓯蓉苯乙醇總苷脂質體。
2.3 苯乙醇總苷脂質體的體外性質考察
2.3.1 不同方法制得的脂質體的粒徑、電位測定采用Malvern粒徑測定儀測定脂質體的粒徑和電位,結果見表2,由結果可知,3種方法形成的脂質體粒徑均在200 nm左右,粒徑均勻,電位均在35 ~ 45 mV。
2.3.2 脂質體的包封率測定1)洗脫曲線測定:采用葡聚糖凝膠柱洗脫法測定脂質體的包封率,取SephadexG-50裝柱,使徑高比為1∶10(cm∶cm),取1 mL脂質體上樣,用pH 7.2的磷酸鹽緩沖液洗脫,流速控制在1 mL·min-1,每2 mL接取一個流份,共接取30個流份,所得流份分別用2 mL甲醇溶解,置于330 nm處測定吸收度,以流份為橫坐標,以藥物濃度為縱坐標,繪制洗脫曲線。結果表明,脂質體流份在第3 ~ 6流份流出,游離藥物在7 ~ 14流份流出。
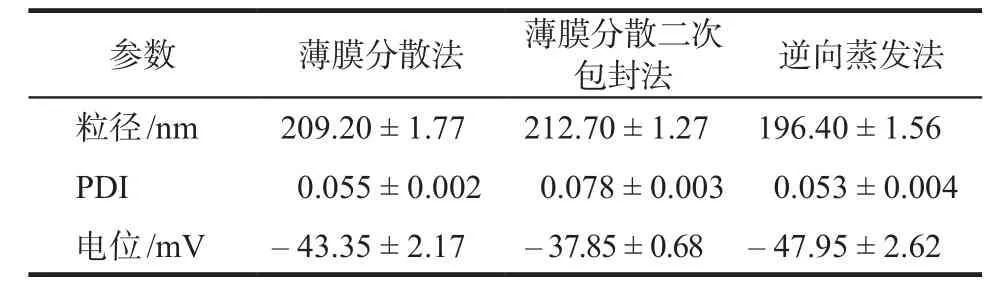
表2 不同方法制得的肉蓯蓉苯乙醇總苷脂質體的粒徑. n = 3Tab 2 Particle size of the phenylethanoid glycosides liposomes prepared by different methods. n = 3
2)不同制備方法制得的脂質體包封率的測定:包封率測定方法為:取SephadexG-50(粒徑100 ~ 300 μm)裝柱,使徑高比為1 cm∶10 cm,取不同制備方法制得的脂質體1 mL分別上樣,用pH 7.2的磷酸鹽緩沖液進行洗脫,流速控制在1 mL·min-1,收集脂質體流份,從中取2 mL加甲醇破乳溶解后測定松果菊苷的含量,作為脂質體中包封的藥物含量。另取苯乙醇總苷脂質體1 mL直接加甲醇破乳溶解后,測定松果菊苷的含量作為脂質體中藥物總量。按下述公式計算包封率:包封率%=(脂質體中包封藥物量/脂質體中藥物總量)×100%。
本研究結果表明,采用薄膜分散法、二次包封法、逆相蒸發法制得脂質體的載藥量分別為(28.55 ±5.61)%,(38.46 ± 7.85)%,(33.88 ± 3.50)%。用SPSS 16.0軟件中SNK-Q檢驗對三種方法制備的肉蓯蓉總苷脂質體包封率均數兩兩比較,以P < 0.05為差異具有統計學意義。結果顯示,二次包封法制得的脂質體的包封率最高。
2.3.3 脂質體的載藥量測定取制得的脂質體1 mL精確稱重,記錄載藥脂質體的總重量,將此1 mL脂質體同“2.3.2”項下方法上樣洗脫后,測得脂質體中的載藥量,依據公式:載藥量=Wd/WL×100%,其中Wd表示包封于脂質體中的藥量,WL表示載藥脂質體的總重量。
結果顯示采用薄膜分散法、二次包封法、逆相蒸發法制得脂質體的載藥量分別為(2.73±0.57)%,(3.71±0.32)%,(3.28±0.49)%。
2.3.4 pPB修飾的肉蓯蓉苯乙醇總苷脂質體的體外性質測定1)pPB修飾的苯乙醇總苷脂質體的粒徑、電位、包封率、載藥量的測定:對不同制備方法所得的脂質體的粒徑、電位、包封率和載藥量的結果進行比較,結果表明,二次包封法制得的脂質體的包封率和載藥量最高,粒徑和電位與其它制備方法相似,因此確定二次包封法是制備脂質體的最優方法。在此基礎上,采用二次包封法制備了pPB修飾的肉蓯蓉苯乙醇總苷脂質體,其粒徑為(227.60±1.63)nm、電位為(- 45.37±1.62) mV、包封率為(35.55±7.35)%、載藥量為(3.80±0.28)%。
2)脂質體形態觀察:取二次包封法制得的p PB修飾的苯乙醇總苷脂質體混懸液1滴,滴至專用銅網上,用磷鎢酸染色后,自然晾干,于透射電鏡下觀察脂質體形態,結果見圖1。

圖1 p PB修飾的苯乙醇總苷脂質體的透射電鏡圖Fig 1 TEM photo of pPB-modified phenylethanoid glycosides liposomes
3 討論
脂質體是磷脂雙分子層構成的囊泡,無毒副作用和免疫原性,用其包封藥物后,具有使藥物生物利用度提高,增加藥物靶向性,降低藥物毒性等優點。同時在脂質體表面偶聯能與靶點高效結合的配體(如pPB環肽)后,可使脂質體與靶細胞表面相應的受體分子特異性結合,從而將藥物靶向至具有特異性受體的細胞、組織或器官。
通過HPLC方法測定,發現肉蓯蓉總苷主要成分為松果菊苷和毛蕊花糖苷,這兩種成分在HPLC條件下能夠同時出峰,其中松果菊苷含量更高,同時根據文獻報道,肉蓯蓉總苷可選用松果菊苷作為指標性成分,因此本研究選用松果菊苷作為測定肉蓯蓉總苷的指標性成分。有文獻報道,除HPLC方法外,由于松果菊苷在總成分中占比較高,因此為了便于測定,本文在采用UV法測定肉蓯蓉苯乙醇總苷含量時,以測定松果菊苷含量為主要指標[14]。
凝膠柱層析法是利用分子篩的原理,通過在凝膠柱上滯留時間的差別對傳遞體和游離藥物進行分離,脂質體的分子大,滯留時間短,洗脫較快;游離藥物分子小,滯留時間長,洗脫較慢,從而使脂質體與游離藥物得到較好的分離。選擇適當的有機溶劑和油水比可制備較穩定的脂質體,本研究過程中發現氯仿和甲醇混合溶劑為有機溶劑,比單獨使用一種溶劑更容易形成穩定的脂質體。
本實驗測得的肉蓯蓉苯乙醇總苷脂質體的包封率較低,可能與該藥物本身的性質有直接的關系。當藥物油水分布系數(logP)< - 0.3或> 4.5時,藥物可被包封成具有較高包封率的脂質體,否則包封率較低。肉蓯蓉苯乙醇總苷的logP約為1,介于兩者之間,這可能是導致脂質體包封率較低的主要因素[15]。
