α-二氧化錳對鉀離子吸附性能的研究及計算
2019-01-16 12:11:20張冬昊黃雪莉
無機鹽工業 2019年1期
關鍵詞:改性
張冬昊,黃雪莉,劉 娜
(新疆煤炭潔凈轉化與化工過程重點實驗室,新疆大學化學化工學院,新疆烏魯木齊830046)
利用吸附的方式,從低濃度海水或鹽湖鹵水中高效提取鉀一直是各國學者研究的重要領域之一[1-2]。M.Kamatsu[3]在專利中提出鈦氧化物用于海水中K+的吸附;J.Hou等[4]合成的沸石分子篩對海水中K+的吸附量為 54.9 mg/g;石勤等[5]合成的鋇十字沸石分子篩對瑪納斯鹽湖鹵水中K+吸附量為70.1mg/g,對羅布泊鹽湖鹵水中K+的吸附量達85.1 mg/g。
類似于多孔結構的沸石分子篩,二氧化錳離子篩作為離子交換中較具優勢的一種吸附劑[6],近年來逐漸為學者所關注。 自然界中存在α、β、γ、δ、ε和λ 等多種晶型[7-8]的二氧化錳,主要以[MnO6]正八面體為單元,相鄰八面體共角或共棱組成含(1×1)、(1×2)、(2×2)隧道的層狀、立體骨架狀空間結構,各晶型的MnO2均具有不同的選擇吸附作用[9-11]。α-MnO2空間群為 I4/m,有(1×1)和(2×2)空間隧道,其中(2×2)隧道尺寸為 0.46 nm,與一系列水合離子(K+、NH4+、Ba2+等)半徑接近[12],且由于晶格缺陷還存在Mn3+造成電荷失衡,這兩點為α-MnO2吸附K+提供了理論條件。董殿權等[13]合成出隱鉀錳礦型MnO2,并測定了其對堿金屬離子的分配系數由大到小依次為 K+、Rb+、Na+,交換性能隨 pH 增加而增大,對 K+的飽和交換容量達到181.5 mg/g。劉月[14]對幾種二氧化錳晶體和表面結構進行了理論研究,主要涉及鐵磁性和反鐵磁性的計算。但總的來說,大多數α-MnO2合成方法周期較長,吸附量不高,有必要進一步研究,包括α-MnO2吸附K+過程的模擬研究。
本文以水熱法合成α-MnO2離子篩,探究了物料物質的量比、水熱溫度、晶化時間、反應溶液中酸濃度對樣品吸附性能的影響,并基于密度泛函理論采用Material Studio8.0進行了分子模擬計算。
1 實驗部分
1.1 實驗分析設備及主要藥品
采用D8型X射線衍射儀(XRD)對樣品的晶體結構進行測試;釆用VERTEX70型紅外光譜儀(FTIR)對樣品進行測試;采用SU8000型掃描電子顯微鏡(SEM)觀察樣品形貌;采用ESCALAB 250Xi型X射線光電子能譜探究樣品化合價。
高錳酸鉀(KMnO4)、硫酸錳(MnSO4·H2O)、氫氧化鉀(KOH)、氯化鉀(KCl),均為分析純;所用水為去離子水。
1.2 α-MnO2離子篩的制備與改性
α-MnO2離子篩合成方法眾多,溶膠凝膠法多以有機溶劑作為助劑,污染較大;氧化還原沉淀法和水熱合成法條件容易控制,產品晶型較好,操作簡單、無污染且反應均勻,但操作時間相對較長。本文選擇水熱合成法合成α-MnO2離子篩,晶體熟化遵循奧斯瓦爾德熟化機理,生成晶體后在高溫高壓下溶解再重結晶成穩定晶體。以一定比例的MnSO4和KMnO4為原料,充分溶解攪拌后轉移至高溫水熱釜,恒溫條件下靜置晶化一定時間,過濾干燥得到樣品。
α-MnO2離子篩在含K+溶液中合成,K+在晶體結構的空腔中起支撐作用。通過酸處理,可以在不改變結構的同時以H+替換K+所在的位置,達到改性的目的。因此,本文中α-MnO2離子篩的改性采用酸浸法,以一定濃度硝酸浸泡水熱合成的含鉀二氧化錳離子篩一定時間,抽濾并反復沖洗至中性后干燥,得 K+被抽出(2×2)隧道的 H 型 α-MnO2離子篩。
1.3 α-MnO2離子篩的吸附
參考海水和鹽湖鹵水中K+的濃度,配制含K+質量分數為0.5%左右的溶液,準確稱取0.2 g吸附劑置入錐形瓶,加入150 mL含鉀溶液,恒溫水浴振蕩吸附足夠時間后,測上清液中K+濃度。單位質量α-MnO2對K+吸附量的計算公式為:
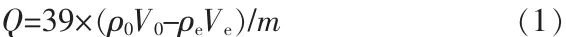
式中,Q表示α-MnO2離子篩對K+的吸附量,mg/g;ρ0、ρe表示初始條件和吸附結束時溶液中K+的質量濃度,g/L;V0、Ve表示初始條件和吸附結束時溶液的體積(體積基本不變),mL;m表示所取吸附劑α-MnO2離子篩質量,g。
2 α-MnO2離子篩的模擬計算研究
2.1 模型構建
采用無機晶體結構數據庫(ICSD)提供的α-MnO2離子篩晶胞結構參數和原子位置,空間群為I4/m,晶胞參數為 a=b=0.978 5 nm,c=0.286 3 nm,α=β=γ=90°。 在 Material Studio8.0 軟件中,以 2×2×2 超胞作為局域周期性晶體模型,晶胞模型的建立和優化采用CASTEP模塊,利用Materials Visualizer建立溶劑模型,采用Forcite模塊對模型進行優化。
2.2 模擬方法
為簡化計算,本文僅以純α-MnO2晶體為例進行計算討論。運用DFT平面波贗勢法,進行第一性原理計算。交互相關函數選擇PBE/GGA(廣義梯度近似函數),為克服廣義GGA函數的局限性比如帶隙值偏小的問題,庫倫校正系數U為2.6 eV。同時考慮到計算的有效性和精確性,k-points設置為fine、截斷能為340 eV時幾何機構和力的優化過程能很好的收斂,費米能級面擴展中高斯拖尾效應為0.1 eV。吸附過程的性質通過巨正則蒙特卡洛(GCMC)方法模擬,采用Sorption模塊,計算任務為Locate,計算方法選擇 Metropolis,平衡步數為 1×108,生產步數為1×105,溫度為25℃。為適應MnO2周期晶體結構,計算采用周期性邊界條件,力場為COMPASSⅡ,用密度泛函理論計算離子篩原子電荷,靜電勢能和Vander Waals勢能分別采用Ewald加和方法和Atom based方法。
3 結果與討論
3.1 α-MnO2離子篩的表征
對初始樣品、改性后樣品、吸附后樣品進行FT-IR、XRD、SEM和XPS表征后,從晶體結構、形貌及價態等方面綜合分析離子篩。一系列樣品的結果對比見圖1~3。所選樣品初始合成條件為:n(K)∶n(Mn)=1∶2、反應溫度為 90 ℃、酸濃度為 1 mol/L、反應時間為14 h。
二氧化錳離子篩FT-IR表征結果見圖1a。圖1a所示結果與文獻[15]中α-MnO2晶體譜圖吻合,3366、3 373、3 392、1 619、1 621、1 384、1 115、960 cm-1為H2O或H3O+的伸縮振動吸收特征峰,也就是晶體孔道內水,而 708~715 cm-1、518~526 cm-1和 466~470 cm-1為[MnO6]正八面體框架中Mn—O鍵的伸縮振動吸收峰,與α-MnO2的FT-IR波譜數據一致。如圖1a所示,經過改性和吸附改性后樣品在1 384、1 115、960 cm-1處的吸收峰消失,推測為改性樣品結構中暴露出的—OH與 H+或 K+結合成 H3O+或K+·H2O,以至于樣品部分吸收峰消失。樣品證明為所需的α-MnO2離子篩,并且在改性后及吸附后樣品的晶體固有性質沒有改變。
合成樣品XRD表征如圖1b所示。改性前后與吸附后晶體的出峰位置大致不變,與PDF(JCPDS 44-1404)標準卡片的出峰位置和強度基本吻合,沒有其他雜峰出現,證明為四方相I4/m空間群的α-MnO2晶體。經過對XRD表征曲線的計算,單元晶胞為a=b=0.978 91 nm、c=0.285 66 nm,與標準卡片十分匹配。改性后的α-MnO2峰位強度比改性前明顯增強且尖銳說明晶體純度更高,吸附后在(110)晶面和(200)晶面出峰位置處的峰強略有降低且峰變寬,進一步驗證晶體在吸附K+之后晶胞尺寸變大。
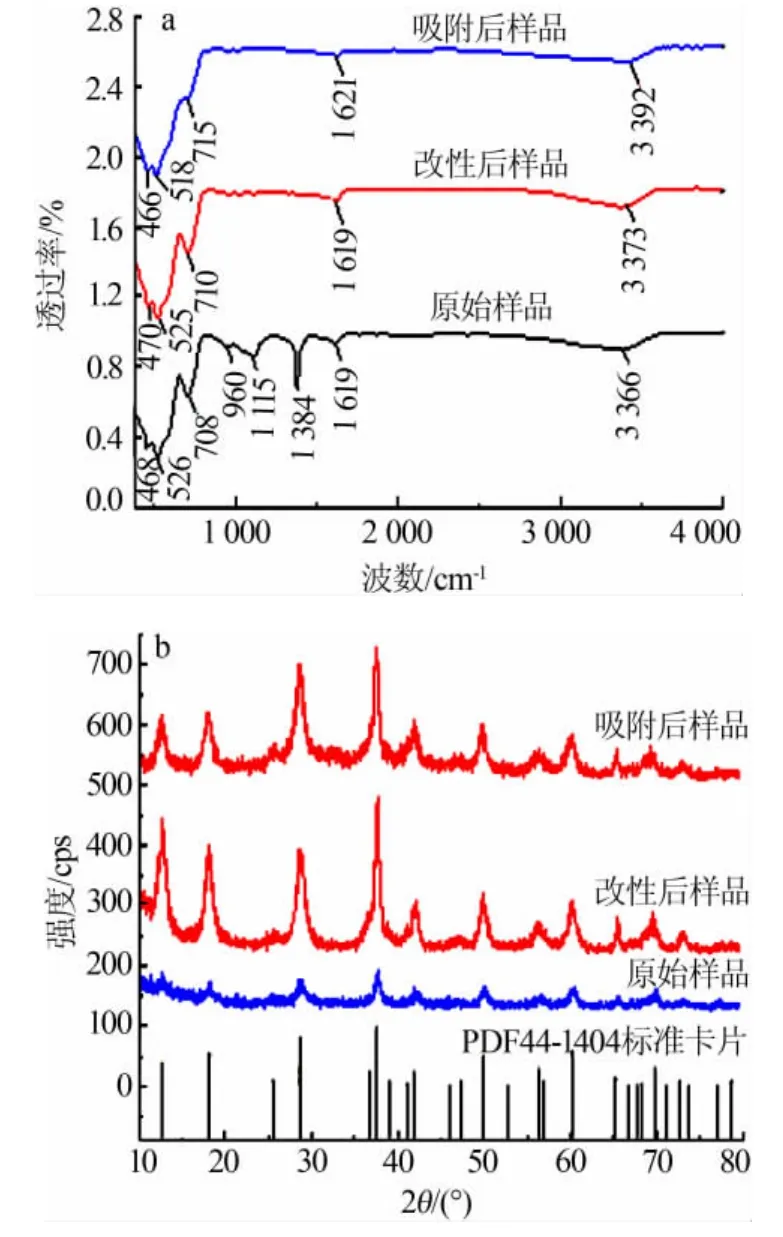
圖 1 樣品的 FT-IR(a)和 XRD(b)表征結果
對改性后的α-MnO2離子篩進行XPS數據分析,結果如圖2所示。圖2a為晶體各主元素的分峰,峰面積之比代表相對含量,圖2a的Mn與O峰面積之比約為1∶2,證明樣品為MnO2。根據MnO2結合能得知,642.7 eV處的Mn2p正是代表了α-MnO2中Mn元素的分峰。同元素不同價態的比較可以通過分析單一元素分峰圖得到。圖2b為Mn2p分峰圖,Mn2p軌道含Mn2p1/2(結合能653.3 eV)和Mn2p3/2(結合能 641.7 eV),分別對應 Mn3+和 Mn4+,通過計算對應 XPS 曲線峰面積可得 n(Mn3+)∶n(Mn4+)約為 1∶1.05。由電荷平衡原理可知,Mn3+的存在使得整體缺正電荷才令離子篩具有陽離子空位,有了吸附K+的電荷基礎,根據 n(Mn3+)/n(Mn4+)可計算得本組樣品最大吸附量為218.67 mg/g。
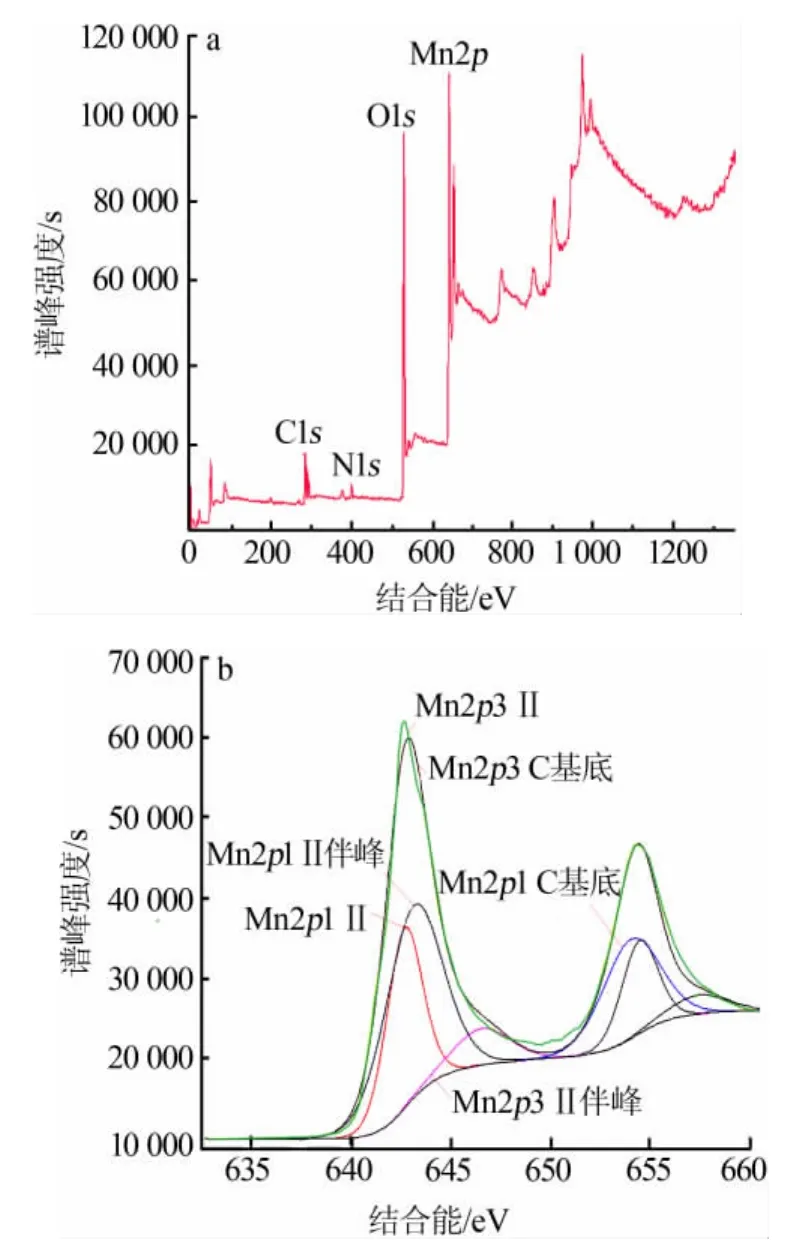
圖2 α-MnO2晶體XPS表征數據圖
合成樣品的SEM形貌如圖3所示。由圖3可見,合成的晶體形貌結構呈矩形條狀或棒狀。由于α-MnO2具有各向異性生長的特性,晶體陳化由晶核為引發點沿不同方向生長成為團簇,在水熱條件下形成均勻I4/m四方相方形斷面的納米棒,并且納米棒團聚成花團。但經酸改性,晶體形貌發生一定的改變,花團狀團簇被破壞,棒狀變為雜亂無章的線狀。推測為H+替代K+位置后,單一晶胞尺寸略縮致使整個形貌有區別,原有的團簇被打亂,呈無序小棒狀。吸附后較吸附前形貌無明顯區別,但分布更為均勻,由改性后的線狀略轉為矩形條狀。

圖3 樣品SEM圖
3.2 α-MnO2離子篩的合成條件對吸附性能的影響
通過單因素實驗,研究了合成條件對吸附劑吸附性能的影響,實驗結果如圖4所示。
從圖4可以看出,鉀的吸附量隨MnSO4用量增加先增加后不變,n(MnSO4)∶n(KMnO4)=3 時最高,為203.57 mg/g;鉀吸附量隨合成溫度升高先增加后減少,110℃時最高,達到214.48 mg/g;鉀吸附量隨硫酸濃度增大先增加后減少,在硫酸濃度為1.5 mol/L時最高,為225.39 mg/g;鉀吸附量在反應時間為12 h時最高,為236.3 mg/g,各組樣品最高吸附量均高于文獻值 181.54 mg/g[13]。
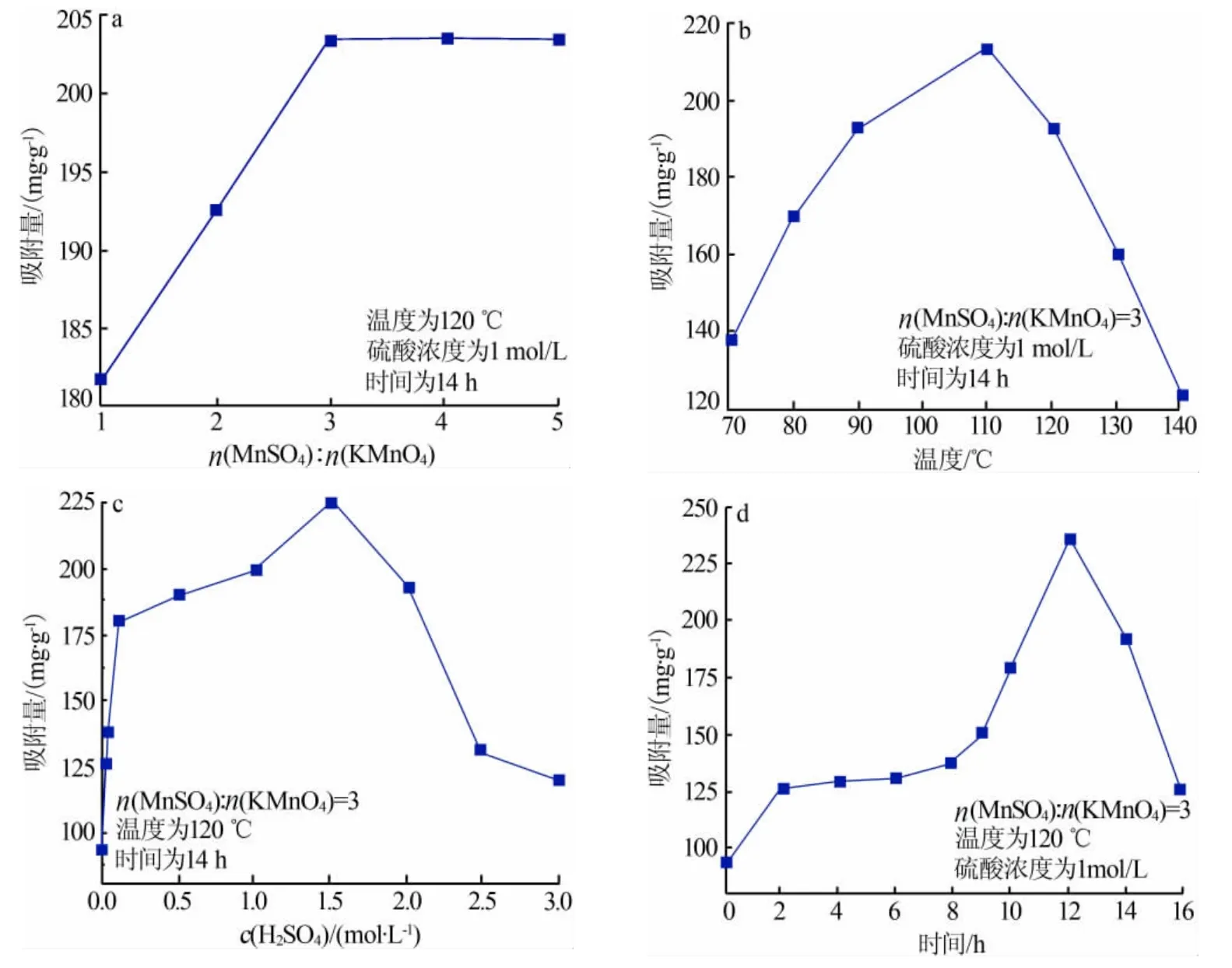
圖4 吸附量隨4個因素的變化
吸附完成后,需要對吸附劑中的鉀進行脫附,以獲得鉀的富集液。為了將吸附劑結構中K+抽出,使其能夠盡可能轉為H型α-MnO2,必須要對樣品進行酸處理。經過酸處理后的樣品需要用去離子水沖洗,直至洗滌后的上層清液呈中性。酸處理過后以無水乙醇和蒸餾水交替洗滌,不需多次洗滌干燥樣品,即可快速達到洗滌目的。被選樣品的循環實驗結果見圖5,在5次吸脫附循環后,樣品的吸附量由214.48 mg/g降至181.75 mg/g,然后保持不變,證明該樣品有著優異的吸附性能和再生性能。
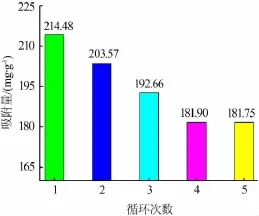
圖5 吸附量隨循環次數的變化
3.3 α-MnO2離子篩的模擬計算研究
理論計算值空間群為I4/m,晶胞參數為a=b=0.978 5 nm、c=0.286 3 nm,實驗結果值空間群為I4/m,晶胞參數為a=b=0.974 93 nm、c=0.283 76 nm,實驗值與理論晶胞參數相符。通過對離子篩吸附構型和相應的吸附性能進行計算比較,對吸附能、吸附位及最大理論吸附量等進行分析。計算得吸附能ΔH為-0.030 7 kJ/mol,證明吸附反應為放熱過程,因此α-MnO2適合在高溫下脫附、低溫下吸附。經Sorption模塊計算,吸附位為離子篩(2×2)隧道,飽和吸附示意圖如圖6所示,最大理論吸附量為299.62 mg/g。通過對比計算值和實驗值發現,實驗測得最大吸附量為計算值的78.87%,仍有較大的提升空間。
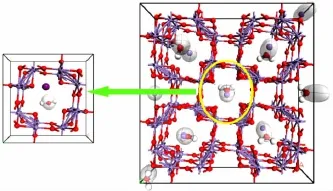
圖6 吸附位及飽和吸附示意圖
4 結論
通過對 FT-IR、XRD、SEM、XPS 結果分析后得知,制得高純度納米棒 α-MnO2[n(Mn3+)∶n(Mn4+)接近1∶1.05]的最優合成條件為:n(MnSO4)∶n(KMnO4)=3、反應溫度為110℃、硫酸濃度為1.5 mol/L,反應時間為12 h。樣品有著較好的再生性能和吸附性能,最高吸附量為236.3 mg/g,是最大理論吸附量的78.87%。吸附位為離子篩(2×2)隧道,吸附反應為放熱過程,低溫更適合吸附過程的進行。
猜你喜歡
紡織科學研究(2020年1期)2020-05-21 00:31:06
中國塑料(2016年12期)2016-06-15 20:30:07
中國塑料(2016年2期)2016-06-15 20:30:00
中國塑料(2016年2期)2016-06-15 20:29:59
中國塑料(2016年5期)2016-04-16 05:25:36
廣西林業科學(2016年3期)2016-03-16 05:43:30
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
中國塑料(2015年4期)2015-10-14 01:09:19
