位阻效應對受阻酚雜化體系阻尼機理的影響
2019-03-19 09:21:12胡喬曼徐康茗
原子與分子物理學報 2019年1期
關鍵詞:體系
胡喬曼, 徐康茗
(1. 重慶文理學院 新材料技術研究院,重慶,402160; 2.重慶文理學院 材料與化工學院,重慶,402160)
1 引 言
隨著城市化進程的不斷加快以及高新技術的迅速發展,機械設備等帶來的振動問題越發突出并已成為城市發展的制約因素.為了減少振動污染,人們致力于研究振動的控制方法,其中聚合物基阻尼材料因減振性能最為突出而被廣泛應用.然而傳統的聚合物基阻尼材料(共混、共聚以及IPN結構)以自身黏彈阻尼機理為主,僅通過分子鏈內摩擦耗散能量,對阻尼性能提升能力有限,已漸漸難以滿足設備等對阻尼材料越來越高的要求.因此,如何研究新的阻尼機理、制備新型阻尼材料已成為減振領域急需突破的關鍵技術問題.
利用動態氫鍵相互作用增強能量耗散制備阻尼材料,相比于傳統阻尼材料,其在動態外力作用下,氫鍵發生可逆的斷裂和重生,吸收大量外部能量,同時引起分子鏈運動加劇、增大內耗,因此成為極具潛力的阻尼改性機理[1]. 該阻尼機理由吳馳飛等人在研究氯化聚乙烯(CPE)/受阻酚雜化阻尼材料時首次提出[2, 3],且因阻尼改性效果顯著,針對該機理的理論研究其后在其他聚合物/受阻酚體系中被廣泛討論.如張立群課題組首次結合分子動力學模擬方法[4],從分子角度探討了丁腈橡膠(NBR)/受阻酚AO-80雜化體系的阻尼機理,模擬結果表明分子間氫鍵數目多少,結合能及自由體積大小是阻尼性能高低的決定因素.其后,研究者們在對聚醋酸乙烯酯(PVAc)/AO-70二元體系[5]及NBR/聚氯乙烯(PVC)/AO-80三元體系[6]的模擬研究中,得到了相似的結論.同時郭少云課題組在熱塑性聚氨酯彈性體(TPU)/受阻酚AO-70雜化體系的模擬研究中進一步發現,分子間氫鍵相互作用種類也是影響阻尼性能高低的重要因素[7]. 由此可見,分子動力學模擬方法可有效地對聚合物/受阻酚體系阻尼機理進行分析研究,而且分子動力學模擬方法在其它研究領域也得到了廣泛的應用[8, 9],證明此方法是一種非常有效地表征手段. 然而值得注意的是,目前研究者們主要集中于探討聚合物結構變化對雜化體系阻尼性能的影響,對雜化體系中另一重要組成單元,受阻酚結構變化與阻尼性能間關系的系統理論研究卻鮮有關注.
針對這一問題,本文以已有實驗研究報道為參考[10],利用分子動力學模擬方法,構建了不同受阻程度受阻酚分子/PVAc雜化體系,重點從理論角度探討受阻程度與雜化體系阻尼性能間的相互關系,并期望相應的數據能更好地為制備新型雜化阻尼材料提供理論支持.
2 分子模型及模擬方法
2.1 分子模型
本文使用的計算軟件是Accelrys的Materials Studio 7.0.首先應用Visualizer模塊構建受阻程度由小到大的TDP、AO300及TBBP三種受阻酚分子模型,相應的分子結構如圖1所示. 接著參考我們的前期工作[5],構建了一條重復單元數為50的PVAc大分子鏈.在對受阻酚及PVAc進行優化得到穩定分子結構后(具體優化步驟見1.2),運用Amorphous Cell模塊構建了以兩條PVAc大分子鏈為基體的,受阻酚重量分數依次為0、10、20、40、60、80的具有周期性邊界的無規盒子,以備后續模擬分析,不同無規盒子中受阻酚添加量如表1所示.
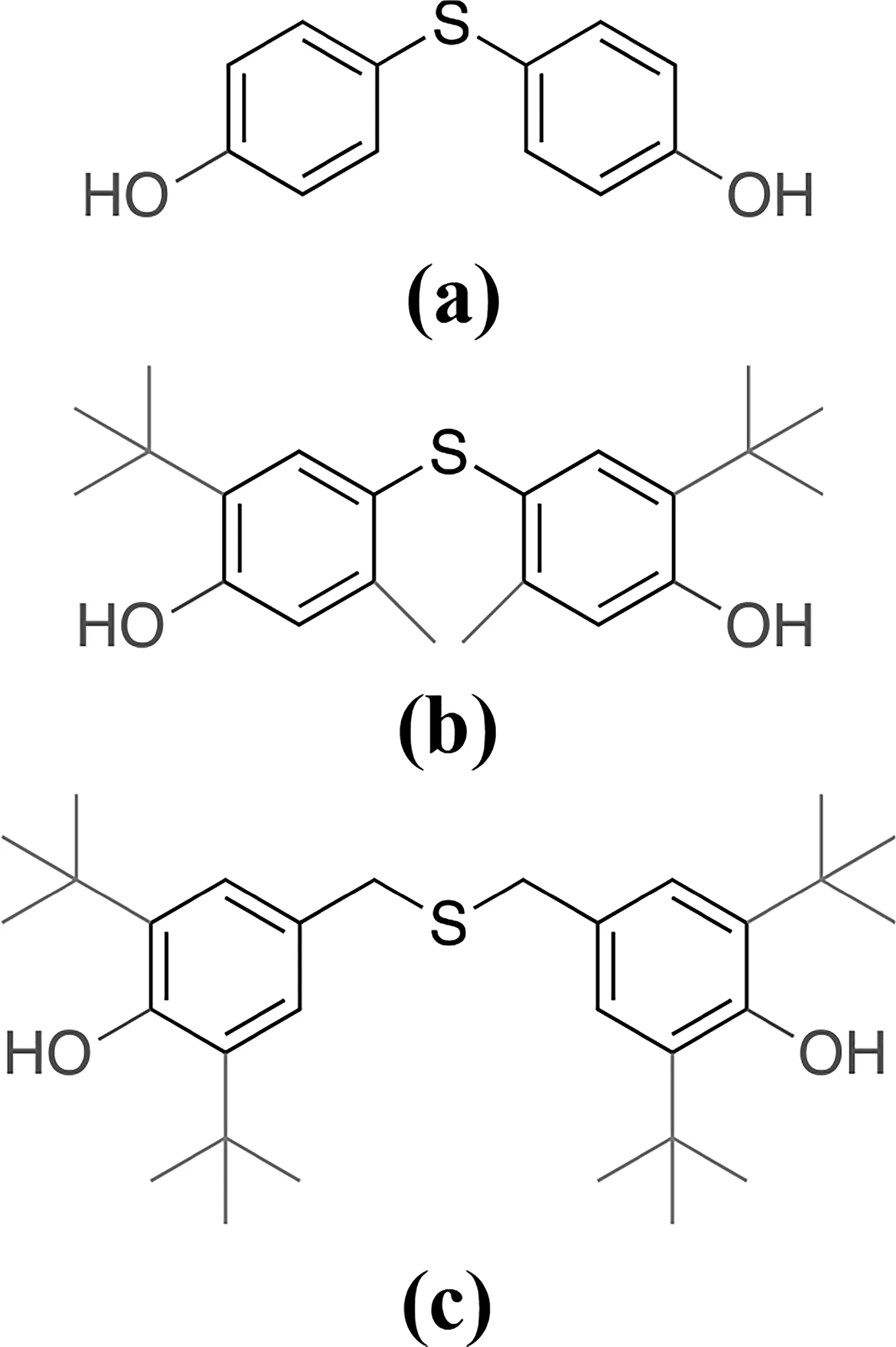
圖1 分子模擬中三種不同受阻酚化學結構式(a)TDP;(b)AO300;(c)TBBPFig.1 Chemical structures of (a) TDP, (b) AO300 and (c) TBBP used in the simulation
Table 1 Adding numbers of hindered phenols in different amorphous cells
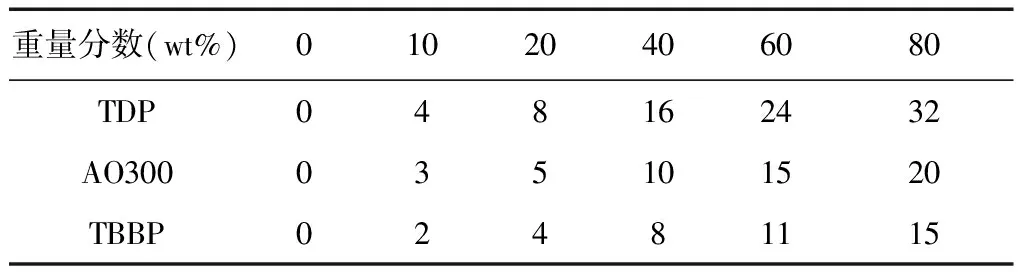
重量分數(wt%)01020406080TDP048162432AO300035101520TBBP02481115
2.2 模擬方法
模擬溫度為25 °C,模擬力場為COMPASS力場,COMPASS力場被廣泛用于優化及預測聚合物結構、構型構象、聚集態性能以及分子間相互作用等研究[11].初始速度采用麥克斯韋-玻耳茲曼分布方法在25 °C下得到,verlet速度時間積分法時間步長為1 fs[12].
圖2為分子動力學模擬流程:運用Forcite模塊對構建的受阻酚及PVAc分子鏈依次采用最速下降法、共軛梯度法及牛頓方法進行幾何結構優化,在此基礎上進行動力學優化,系綜采用NVT,動力學結構松弛時間總長為2 ns,以得到結構穩定的受阻酚及PVAc分子鏈 [圖2(a)、(b)].根據配比分別構建好相應無規盒子[圖2(c)]. 然后對無規盒子進行能量優化,能量收斂方式依次采用最速下降法和共軛梯度法及牛頓方法,接著為了得到幾何尺寸最佳以及能量最低的無規盒子,對無規盒子進行動力學模擬,模擬所用系綜為NPT系綜,所用壓力為1 atm,控溫方法為Anderden, 控壓方法為Berendsen, 動力學結構松弛時間總長為2 ns,其中每隔 5 ps收集一次數據,并采用最后1 ns數據進行結果分析[圖2(d)].最后將經過以上優化過程且能量穩定的無規盒子進行分析.采用軟件自帶氫鍵計算語言計算體系中氫鍵個數 [圖2(e)], 采用軟件自帶分析部件計算體系自由體積以及結合能[圖2(f)].自由體積計算采用網格掃描方法.

圖2 PVAc/TDP分子動力學模擬模型(紅色原子是氧原子,綠色原子是氫原子,灰色原子是碳原子,黃色原子是硫原子,藍色虛線代表氫鍵)Fig. 2 Models for MD simulation of PVAc/TDP hybrids (red atom is O, green atom is H, grey atom is C, yellow atom is S and blue dashed line represents H-bonds)
3 結果討論
3.1 氫鍵相互作用
配對相關函數g(r)可反映以特定原子為球心,在距離r范圍內另一原子出現的概率大小,故而被廣泛用于研究聚合物體系是否存在氫鍵相互作用.兩原子間距離處于2.6-3.1 ?,3.1-5 ?及大于5 ?分別表示兩原子間可產生氫鍵,強范德華力及弱范德華力相互作用[13].圖3以PVAc/TDP-10 wt%模擬體系為例,給出了該雜化體系中分子間氧原子與氫原子隨距離改變的配對相關函數.由圖可知,配對相關函數的最大峰位于2-3.1 ?區間,說明氧原子與氫原子有較大概率形成分子間氫鍵相互作用.需要說明的是,其余雜化體系配對相關函數曲線與圖3類似,故本文中不再逐一列出.
基于配對相關函數分析,表2 給出了不同雜化體系、不同類別氫鍵的平均數目.計算方法為分別對經過相同模擬條件的五個能量平衡的無規盒子進行氫鍵計算,然后求平均值.從表中可以看出,隨著受阻酚添加量的增加,受阻程度最小的TDP體系分子間羥基與羰基的氫鍵數目(TDP Hbondinter)先增加,在80 wt%含量時出現略微的減少,分子內羥基與羥基的氫鍵數目(TDP Hbondintra)
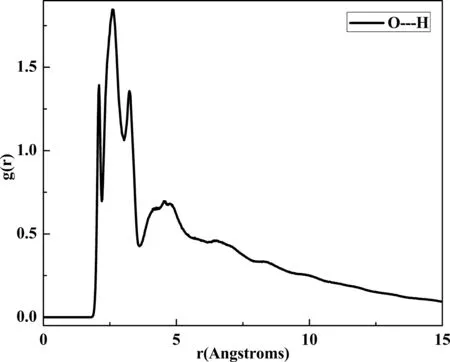
圖3 PVAc/TDP(TDP重量分數10%)混合體系中氫原子和氧原子分子間配對相關函數Fig. 3 Pair correlation function for intermolecular H and O in PVAc/TDP-10 wt% hybrid.
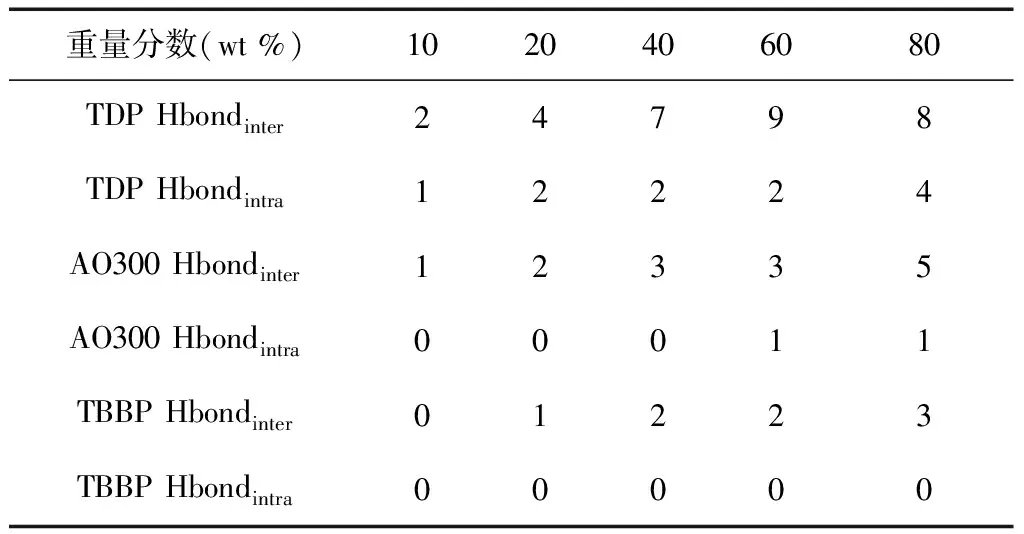
重量分數(wt %)1020406080TDP Hbondinter24798TDP Hbondintra12224AO300 Hbondinter12335AO300 Hbondintra00011TBBP Hbondinter01223TBBP Hbondintra00000
則逐漸增加;受阻程度居中的AO300體系分子間氫鍵數目逐漸增加,分子內氫鍵則在較高含量(60、80 wt%)時才觀測到;受阻程度最大的TBBP體系分子間氫鍵數目在10 wt%體系中未能觀測到,但隨后數目逐漸增加,同時分子內氫鍵也未能在研究體系中觀測到.對上述結果進行比較可知,受阻程度增加,一方面可有效減少受阻酚自身分子內氫鍵相互作用的形成,也即減少小分子自身團聚傾向;另一方面也增大了受阻酚與聚合物間氫鍵相互作用形成的難度.
3.2 結合能
結合能(Ebinding)可反映兩相互作用組分混合程度,其值可通過分子間相互作用能(Einter)求得[14].而Einter可通過平衡狀態下混合體系總能量(Etotal)與單獨組分能量差值求得.以TDP體系為例,Ebinding值可通過公式(1)求得:
Ebinding=-Einter=-(Etotal-ETDP-EPVAc)
(1)
其中,Etotal表示雜化體系總能量;ETDP表示TDP總能量;EPVAc表示PVAc 總能量,由于無規盒子中PVAc分子鏈重復單元數及分子鏈數目是固定的,因此對于所有雜化體系,EPVAc均為一常值,大小為-1929.97 kcal/mol.
表3 不同雜化體系中不同受阻酚含量的結合能
Table 3 Binding energies of different hybrids with different molecular weights
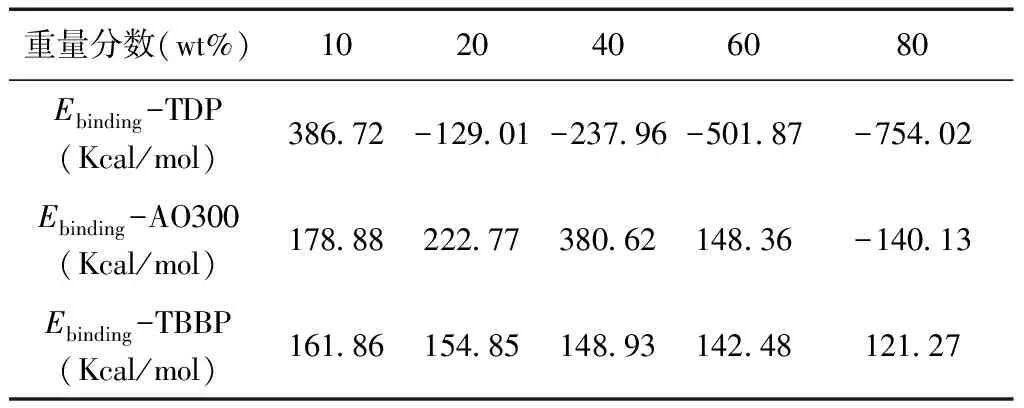
重量分數(wt%)1020406080Ebinding-TDP(Kcal/mol)386.72-129.01-237.96-501.87-754.02Ebinding-AO300(Kcal/mol)178.88222.77380.62148.36-140.13Ebinding-TBBP(Kcal/mol)161.86154.85148.93142.48121.27
圖3為根據公式(1)計算得到的不同受阻酚含量的不同雜化體系的結合能數據.對于TDP體系,受阻酚含量為10 wt%時,其結合能為正值,說明受阻酚與聚合物混合程度較好;繼續增加受阻酚含量,相應體系的結合能則為負值,且含量越大,結合能越低,說明受阻酚與聚合物混合程度越來越差,這主要是由于TDP分子團聚所致.對于AO300體系,隨著受阻酚含量增加,結合能呈現先增加后降低的趨勢,在含量為60 wt%時,也即AO300開始出現分子內氫鍵相互作用時,結合能開始下降,而含量為80 wt%時,結合能變為負值.對于TBBP體系,隨受阻酚含量增加,結合能逐漸降低,但均為正值,說明極性的TBBP能較好地分散在聚合物基體中,但由于位阻作用,兩者間結合相對較弱.
3.3 相對自由體積
根據以自由體積理論推導的Williams-Landel-Ferry (WLF)方程可知[15],聚 合物體系中相對自由體積(FFV)受氫鍵相互作用影響很大.FFV的一般定義如公式(2)所示:
(2)
其中,V和V*分別表示總體積和占有體積.
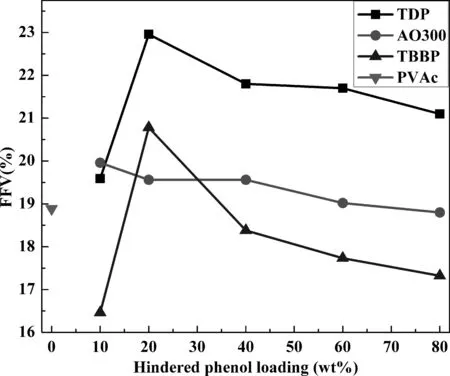
圖4 不同雜化體系中相對自由體積隨受阻酚含量變化曲線Fig. 4 Fractional free volumes of different hybrids.
FFV常用于描述聚合物體系鏈堆積程度大小和自由空間數目的多少. 圖4為不同雜化體系相對自由體積隨受阻酚含量變化曲線. 對于TDP體系,FFV隨受阻酚含量增加呈現先增加后降低的趨勢,且各含量FFV均大于純PVAc體系FFV,這同樣是由于小分子團聚所致. 對于AO300體系,FFV隨受阻酚含量增加而降低,且除了80 wt%體系,其余各含量FFV亦均略大于純PVAc體系. 對于TBBP體系,FFV同樣呈現先增加后降低的趨勢,且除了除了20 wt%體系,其余各含量FFV均小于純PVAc體系,這可能是由于TBBP可較均勻分散于PVAc基體中所致.
3.4 受阻酚擴散系數
圖5為不同雜化體系擴散系數隨受阻酚含量變化曲線.由于各體系相對自由體積均較小,同時受阻酚分子相對較大,圖5中受阻酚擴散系數均較小,說明整個模擬階段,受阻酚僅在小范圍內運動.但值得注意的是,相較于受阻程度最小的TDP及受阻程度最大的TBBP,除極個別情況外,受阻程度居中的AO300擴散系數均更大些,說明小分子團聚及較大的位阻作用均不利于受阻酚小分子擴散,而AO300更大的擴散系數更有利于其與聚合物間形成結合能力更強的分子間氫鍵相互作用.
綜上,受阻程度最小的TDP體系,可形成較多的分子內及分子間氫鍵相互作用,但由于較易形成小分子團聚,其與聚合物混合程度較差、同時自身運動受限導致相對自由體積加大,這些因素均不利于阻尼性能的提高.受阻程度最大的TBBP體系,形成分子間氫鍵相互作用較少且與聚合物間結合較弱,導致其更多地是以普通極性分子的形式分散于聚合物基體中,分子間氫鍵作用效果不明顯,同樣不利于阻尼性能的提高.受阻程度居中的AO300體系,在一定含量下可形成適中的分子間氫鍵相互作用與結合,有利于發揮氫鍵作用提升阻尼性能,但過高含量同樣可導致其形成小分子團聚降低阻尼性能.
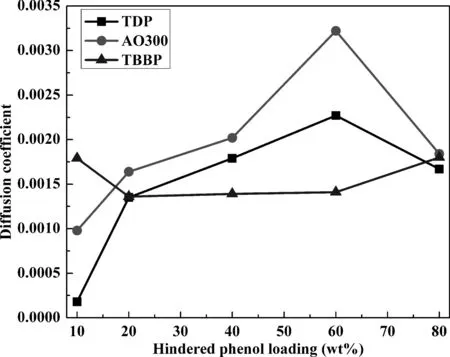
圖5 不同雜化體系中擴散系數隨受阻酚含量變化曲線Fig. 5 Diffusion coefficients of different hybrids
4 結 論
本文通過對受阻程度不同的三種受阻酚小分子/PVAc體系進行分子模擬分析,揭示了位阻效應對雜化體系阻尼機理的影響,主要結論如下:
(1)受阻程度增加,雜化體系分子內及分子間氫鍵相互作用減弱,其中分子內氫鍵相互作用弱化效果更明顯,可有效減少小分子團聚傾向.
(2)小分子團聚及過大的位阻效應均對受阻酚運動產生阻礙作用,不利于受阻酚與聚合物形成強烈氫鍵相互作用,從而使結合能偏低,同時小分子團聚使得相對自由體積較大,過大位阻效應使得氫鍵作用難以發揮,這些均不利于阻尼性能的提高.
(3)合適的位阻既能減小小分子團聚,同時對受阻酚運動阻礙作用較小,可使得其與聚合物基體形成強烈的氫鍵相互作用,利于阻尼性能的提升.
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
