CH4、H2O在CaCO3 (010) 表面吸附的第一性原理研究
2019-03-19 09:21:24趙建飛
原子與分子物理學報 2019年1期
秦 娟, 郭 平 , 趙建飛, 羅 強
(1.西南石油大學油氣藏地質及開發工程國家重點實驗室, 成都 610500; 2.西南石油大學理學院, 成都 610500)
1 引 言
隨著常規油氣資源的不斷減少, 非常規油氣資源的勘探開發已逐步成為各國關注的焦點[1]. 我國致密氣資源豐富, 具有一定的開發技術, 致密氣開發基礎設施建設較為完善, 是我國目前最現實的非常規天然氣資源[2]. 但是對于致密氣藏在儲層中的賦存形式到底是以吸附態存在還是以游離態存在, 很少有人給出理論依據. 致密碳酸鹽巖中含有大量的方解石, 其礦物成分主要是碳酸鈣, 致密氣的主要成分是甲烷, 且在致密碳酸鹽巖中形成的低孔低滲儲集體往往具有復雜的氣水關系[1,3]. 因此,以甲烷和水為對象, 研究其在碳酸鈣表面的吸附能力, 對于解釋致密氣在儲層巖石基質上的賦存狀態以及合理開發具有重要意義.
近年來, 隨著分子模擬理論和計算機技術的進步, 研究者們已經發展出諸如第一性原理方法、 分子動力學方法和蒙特卡羅方法等計算方法來研究材料的性質, 其中, 基于密度泛函理論的第一性原理方法已經成為固體物理、 量子化學和材料科學中的常規研究手段[4, 5]. 從常規油氣開發到非常規油氣開發的發展, 即從微米尺度到納米尺度的發展, 宏觀技術的突破越來越依賴微觀成果, 于是采用新的理論與方法加強微觀研究就顯得尤為重要[6]. 目前, 研究者采用第一性原理方法對固體表面吸附進行了大量研究[7-9], 但是采用第一性原理方法對CaCO3的研究主要是基于選礦工作對方解石與其他含鈣礦物進行浮選, 以及預測礦物與藥劑的作用機理[10-15], 而從油氣與儲層吸附角度對CH4和H2O在碳酸鈣表面吸附的微觀定性研究較少. 同時, 儲層具有復雜的孔隙結構和礦物成分, 與實驗方法相比, 采用計算機模擬研究CH4與H2O的微觀吸附具有無可比擬的優勢. 本文采用基于密度泛函理論的第一性原理方法展開吸附模擬計算, 以CaCO3為吸附劑, 選取CH4和H2O為吸附質, 定性研究CH4和H2O在CaCO3(010) 面高對稱位的吸附穩定性, 并分析不同吸附位上的吸附能、 物理結構變化和電子態密度, 進而得到CH4與H2O在CaCO3(010) 面的吸附規律.
2 模型建立
碳酸鈣礦物在自然界中主要以方解石(calcite)和文石(aragonite)的形式存在, 研究選取的方解石型CaCO3晶體[16], 屬于三方晶系, 空間群為R-3C(No.167,Z=6). CaCO3原胞結構如圖1所示. 初始晶格常數為a=b=0.499 nm,c=1.706 nm,α=β=90°,γ=120°, 優化后晶格常數未變, 只是碳酸根原子位置發生微調, 與實驗值(a=b=0.499 nm,c=1.781 nm)[17]在誤差允許范圍內. 對優化后的原胞截取(010)面并構建厚度為2 nm的真空層, 得到p(1×1)CaCO3(010) 面模型. 對于單個CH4與H2O分子, 采用1.0 nm× 1.0 nm× 1.0 nm的晶格進行優化. 研究CH4和H2O在CaCO3(010) 面吸附時, 考慮到計算量和基底厚度影響, 對體系進行表面弛豫, 即將表層氧原子與氣體分子外的原子固定. 固定非表面原子后的CaCO3(010) 面的物理模型如圖2所示.
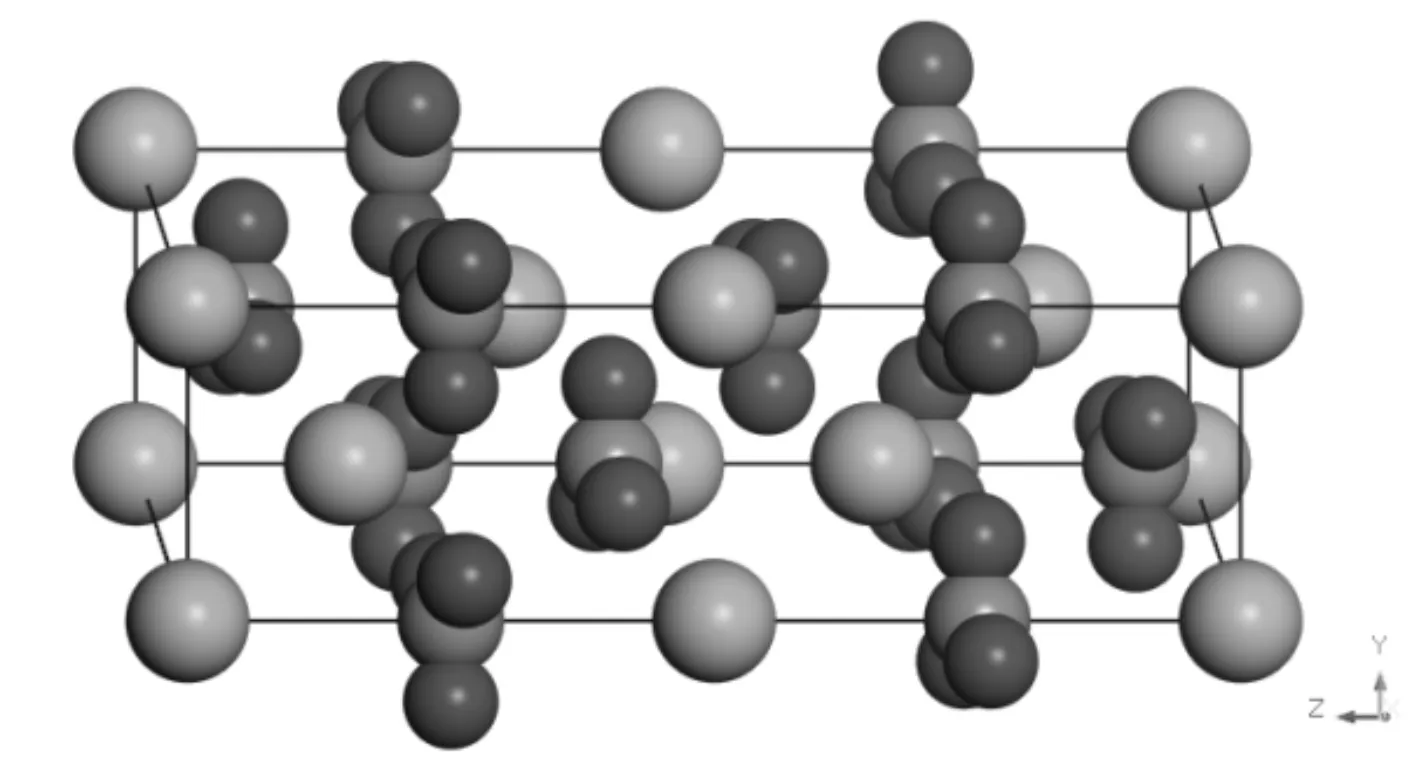
圖1 CaCO3晶胞(紅色: O原子, 綠色:Ca原子, 灰色: C原子)Fig. 1 Crystal cell of CaCO3 (red: O atom, green: Ca atom, gray: C atom)
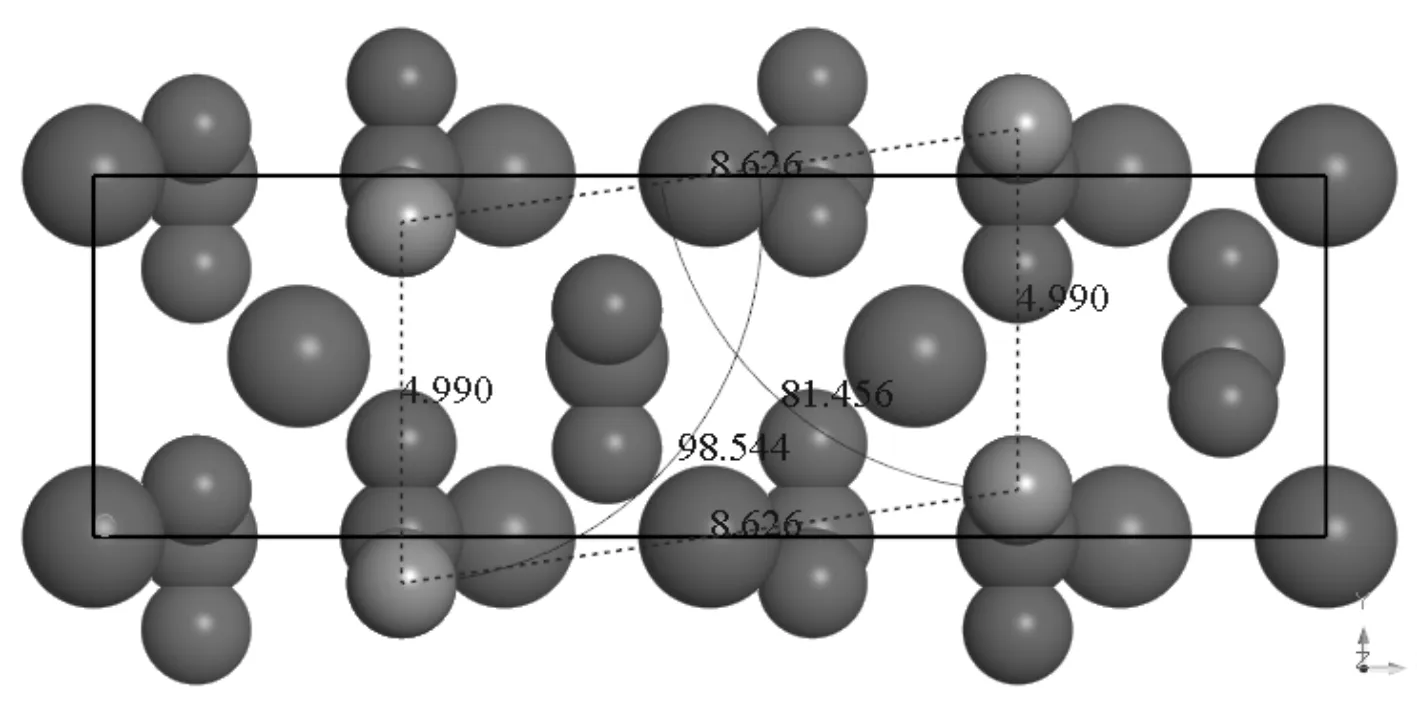
圖2 CaCO3 (010) 面(紅色: 被固定原子, 灰色: 未固定的表層氧原子)Fig. 2 Structural model of CaCO3 (010) surface (red: fixed atom, gray: unfixed atom of O)
最穩定吸附出現在高對稱位上, 而CaCO3(010)模型表面由四個氧原子構成一個平行四邊形, 故存在4個高對稱吸附位. 氧原子正上方為頂位(T位), 平行四邊形的對角線正上方為表面四重洞位(H位), 平行四邊形邊長中點的正上方為橋位(B位), 按邊長不同分為長橋位(LB位)和短橋位(SB位), 如圖3所示. 吸附分子在各吸附位的吸附排列方式按照與表層正對的原子劃分, CH4吸附時有3種空間構型[18]: 1個H原子朝向吸附底物表面而另外3個H原子在上; 2個H原子朝向吸附底物表面而另外2個H原子在上; 3個H原子朝向吸附底物表面而另1個H原子在上, 分別記為Ⅰ、 Ⅱ、 Ⅲ. H2O有1個H原子在底面、 2個H原子和1個O原子在底面的吸附方式, 記為ⅰ、 ⅱ、 ⅲ, 以CH4在H位為例, 吸附方式如圖4所示, 左側為吸附面俯視圖, 右側為正視圖. 計算涉及CH4和H2O兩種分子在四個高對稱吸附位各三種吸附方式, 故有24組高對稱位的吸附.
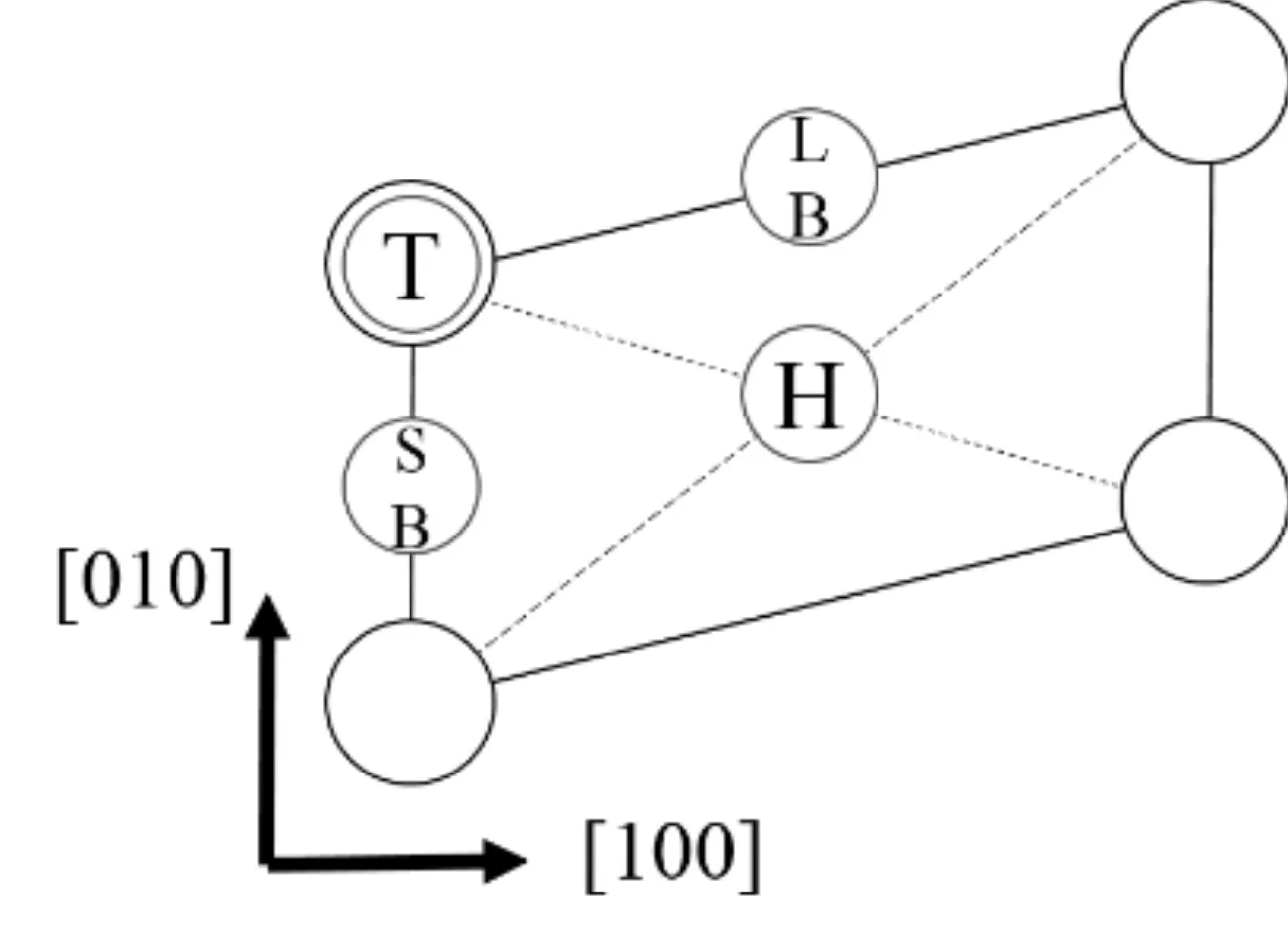
圖3 CaCO3 (010) 面高對稱吸附位: T位、 H位、 LB位和SB位Fig. 3 High symmetry adsorption sites above CaCO3 (010) surface: T site, H site, LB site, and SB site

圖4 CH4在H位的吸附方式: HⅠ、 HⅡ、 HⅢFig. 4 Adsorption directions of CH4 above H site: HⅠ, HⅡ, HⅢ
3 計算方法及過程
4 結果與分析
為了分析CH4和H2O分子分別在CaCO3(010)面吸附的穩定性, 計算了CH4和H2O分子的吸附能Ead, 其公式如下[27]:
Ead=EX/CaCO3(010)-EX-ECaCO3(010)
(1)
其中EX/CaCO3(010)、EX和Ecacoз(010)分別表示CH4和H2O分子分別與CaCO3(010)面吸附體系的總能量、 吸附前單個CH4和H2O分子各自的能量及CaCO3(010)面結構的能量. 吸附能計算值為負時表明發生放熱反應且吸附后體系更加穩定,Ead越小表示體系的穩定性越強; 反之,Ead越大表示體系的穩定性越差. 當吸附能滿足-0.62 eV
4.1 吸附能分析
對以上24組高對稱吸附位的吸附模型進行結構優化計算, 并根據吸附能計算公式得到吸附能, 其結果如表1所示:
表1 CH4和H2O分子在CaCO3(010) 面的吸附能
Table 1 Adsorption energy of CH4和H2O on different adsorption sites above CaCO3(010) surface
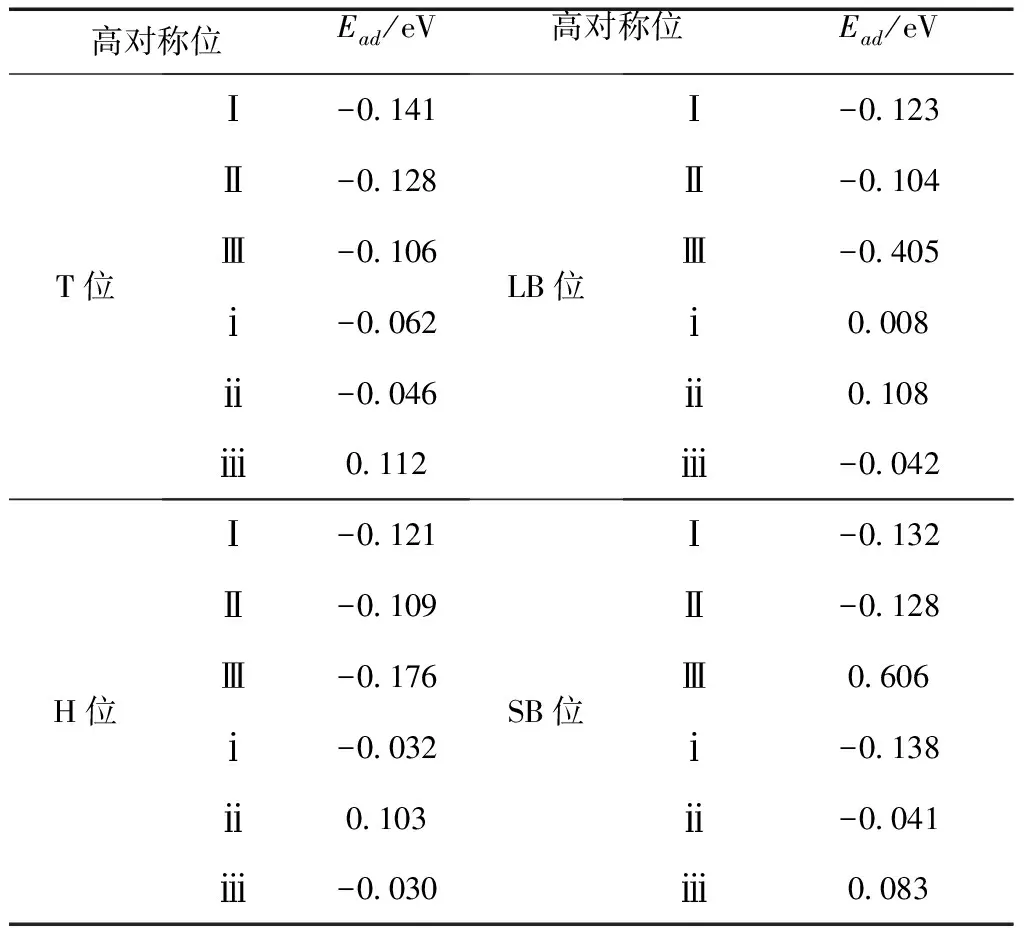
高對稱位Ead/eV高對稱位Ead/eVT位Ⅰ-0.141Ⅱ-0.128Ⅲ-0.106ⅰ-0.062ⅱ-0.046ⅲ0.112LB位Ⅰ-0.123Ⅱ-0.104Ⅲ-0.405ⅰ0.008ⅱ0.108ⅲ-0.042H位Ⅰ-0.121Ⅱ-0.109Ⅲ-0.176ⅰ-0.032ⅱ0.103ⅲ-0.030SB位Ⅰ-0.132Ⅱ-0.128Ⅲ0.606ⅰ-0.138ⅱ-0.041ⅲ0.083
由表1可知, CH4和H2O分子在CaCO3(010)面不同吸附位的吸附能均大于-0.62 eV,吸附不明顯, 為物理吸附. CH4分子在CaCO3(010)面LB位Ⅲ型吸附方式的吸附能最小, 為-0.405 eV, 即CH4分子以三個H原子正對表層O原子時吸附最穩定; 而在SB位Ⅲ吸附方式的吸附能最大, 為0.606 eV, 即CH4分子在SBⅢ位的吸附最不穩定. 從吸附能的變化范圍角度來看, 除吸附最穩定的LBⅢ位與最不穩定的SBⅢ位外, 其他各吸附位的吸附能分布在-0.104 eV到-0.176 eV, 相差較小且為0.072 eV, 表明CH4分子與CaCO3(010)面發生吸附時, 優先在LB位以三個H原子正對表層O原子的吸附方式吸附, 其余吸附方式吸附能力相當, 最后吸附在SBⅢ位. H2O在CaCO3(010)面SB位ⅰ型吸附方式的吸附能最小, 即H2O分子以一個H原子正對表層O原子時吸附最穩定, 吸附能為-0.138 eV, 這表明CaCO3對H2O分子有一定的吸附作用, 可能出現水鎖現象[29]; 而在T位ⅲ型吸附方式的吸附能最大, 為0.112 eV, 即H2O以一個O原子正對表層O原子時吸附最不穩定. 吸附能的變化范圍相差較小, 為0.25 eV. 對比CH4與H2O吸附的吸附能可知, CH4在CaCO3(010)面的吸附性強于H2O在CaCO3(010)面的吸附, 宏觀表現為CaCO3親氣, 與參考文獻[30]所得結論類似.
4.2 物理結構分析
對CH4和H2O各自最穩定吸附的位置吸附前后的物理結構進行分析, 有利于了解吸附作用對流體分子的結構影響, 其吸附前后鍵長、 鍵角的變化見表2:
表2 CH4和H2O分子在 CaCO3(010)面吸附后的鍵長、 鍵角變化
Table.2 Changes of bond length and bond angle of CH4and H2O molecules above the CaCO3(010) surface after adsorption.
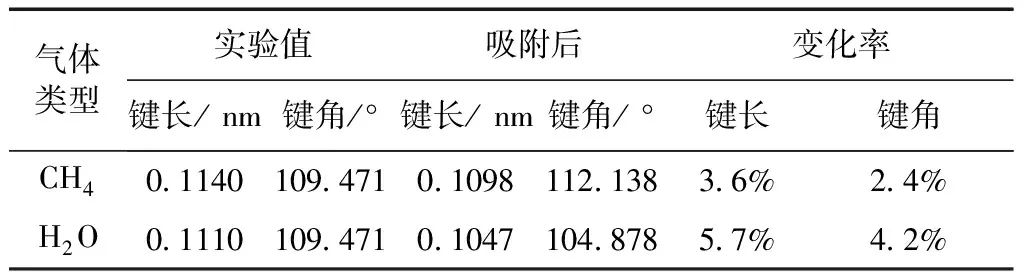
氣體類型實驗值吸附后變化率鍵長/ nm鍵角/°鍵長/ nm鍵角/ °鍵長鍵角CH40.1140109.4710.1098112.1383.6%2.4%H2O0.1110109.4710.1047104.8785.7%4.2%
從表2可以看出CH4分子在CaCO3(010) 面LBⅢ位吸附體系進行結構優化后, 吸附后C-H、 H-O鍵鍵長分別縮短了0.0042 nm、 0.013 nm, 變化率分別為3.6%和5.7%; 吸附后H-C-H鍵角增大了2.6673°, 變化為2.4%, 而H-O-H鍵角變小了4.593°, 變化率為4.2%. 根據上述數據分析可以發現, 鍵長鍵角的變化較小, 表明吸附作用對CH4和H2O分子結構的影響較小.
四是嚴肅對待評議結果的反饋。人大代表對旁聽案件的評議,是人大代表依法監督的實實在在的體現,必須高度重視、認真對待。為此,市人大常委會內務司法與法制工作委員會對收集、整理的代表意見和建議,區分不同情況,采取不同方式反饋給法院,要求法院在規定的時間內及時向人大報告意見和建議的具體落實情況。同時,將人大代表的意見和建議納入法院的內部考評,使代表的聲音真真切切地發揮作用。
4.3 態密度分析
利用CASTEP模塊的Analysis功能可以對優化后的模型進行能帶結構、 電子態密度、光學性質、 聲子色散關系、 聲子態密度以及應力的分析, 此處主要討論吸附極端情況與最穩定吸附前后電子態密度變化及其對吸附作用的影響. 根據吸附穩定性最差與最好的態密度變化的分析, 可以進一步理解吸附能差異的原因, 而CH4和H2O與CaCO3(010)面最穩定吸附前后電子態密度變化可以深入理解吸附作用的強弱.
(1)吸附極端情況體系的態密度
從圖5(a)可看出, SBⅢ位與LBⅢ位的態密度存在一定差異, LBⅢ位態密度曲線峰值整體較SBⅢ大, 能量范圍也較SBⅢ位態密度稍大, 也表明了CH4在LBⅢ位更穩定, 態密度曲線范圍與峰值差異可能與兩體系吸附能差異有關, 源于吸附時吸附方式及原子內電子軌道分布差異.
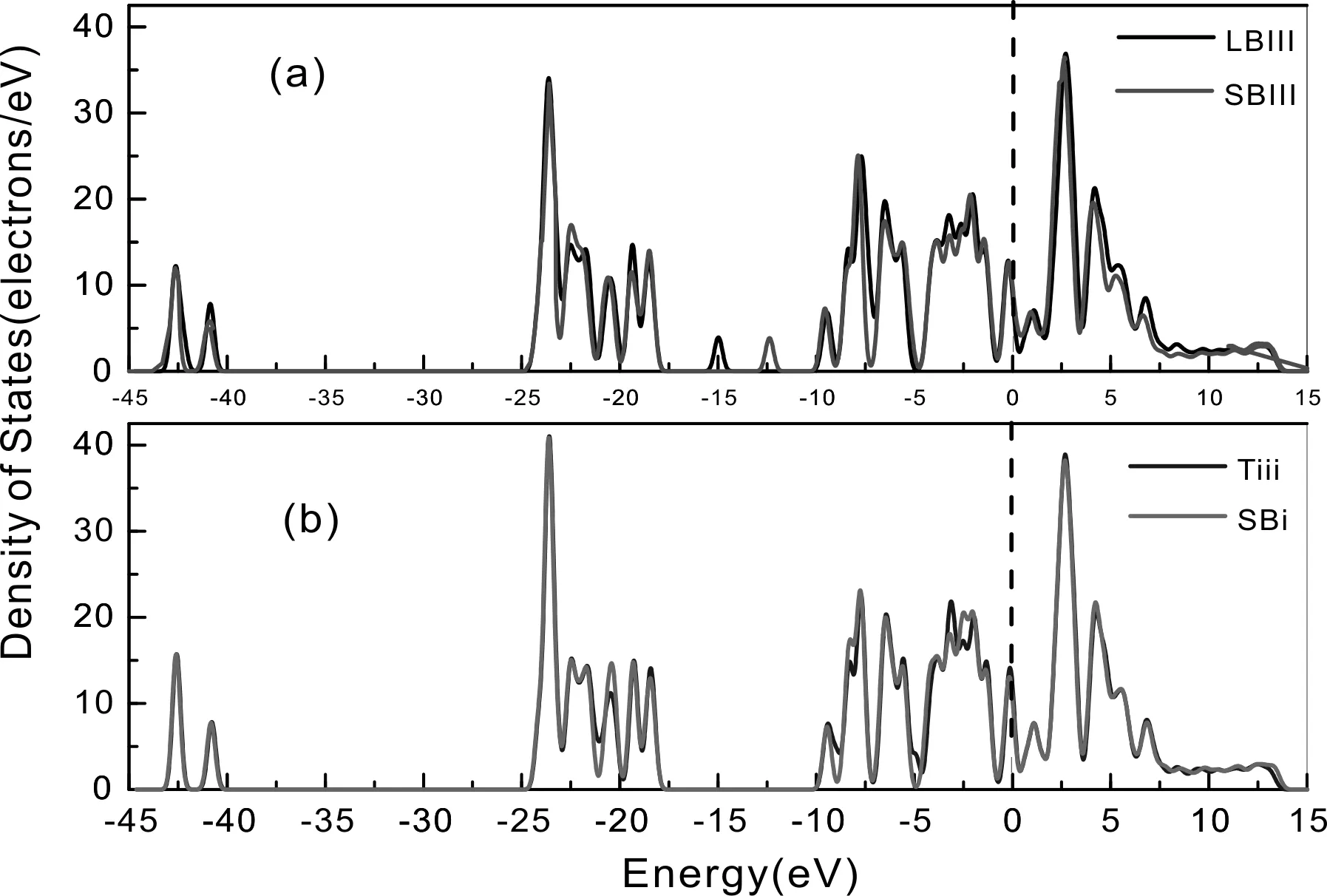
圖5 吸附極端情況體系的態密度Fig. 5 Density of states of extreme adsorption system
從圖5(b)可以看出, Tiii位與SBi位的態密度僅在-22 eV ~ -20 eV、 -9.5 eV ~ -7.5 eV、-56 eV ~ -7 eV和-3 eV ~ -2 eV處存在微小差異, 其余部分基本重合, SBi位態密度曲線在峰值與峰谷處均小于Tiii位態密度峰值, 即SBi位態密度曲線穩定性優于Tiii位, 對應吸附能關系為SBi位小于Tiii位, 也表明了H2O在SBi位吸附更穩定, 同時, 態密度差異性小與吸附能變化范圍小相對應.
(2)吸附前后CH4的態密度
為更好的分析CH4在LBⅢ位吸附前后的電子態密度變化, 將吸附前后CH4的總態密度、s和p分態密度繪制在圖中進行對比分析, 如圖6所示, 能量為0 eV處的虛線表示費米能級[19].
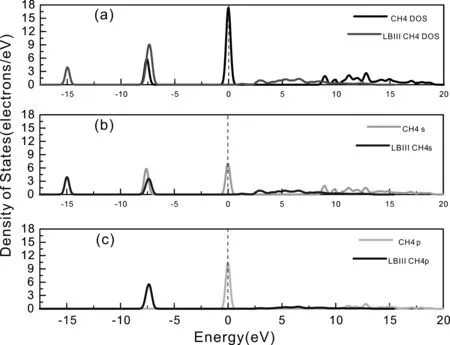
圖6 LBⅢ位吸附前后CH4的態密度Fig. 6 Densities of states of CH4 before and after adsorption on LBⅢ site
圖6(a)為吸附前后CH4的總態密度, 圖中黑色曲線為吸附前CH4的總態密度, 紅色曲線為吸附后CH4的總態密度. 從圖中可以看出, 吸附后的態密度曲線整體向低能量區移動約7.5 eV, 吸附前, 能量分布在-8 eV ~ -7 eV、 -0.75 eV ~ 0.75 eV 與8.5 eV ~ 19 eV區間, 吸附后, 能量分布在-15.5 eV ~ -14.5 eV、 -8 eV ~ -6.5 eV和2.5 eV ~ 12.5 eV區間. 吸附前CH4態密度曲線有2個超過6.0 electrons/eV的峰值且極大值約為17.3 electrons/eV, 而吸附后2個峰值分別約為4.0 electrons/eV、 9.0 electrons/eV, 分別降低約2.0 electrons/eV和8.3 electrons/eV, 這表明吸附后能量降低, 結構更加穩定, 與吸附能為負相對應.
圖6(b)為吸附前后CH4的s分態密度圖, 圖中綠色曲線為吸附前CH4的s分態密度,藍色曲線為吸附后CH4的s分態密度. 從圖中可以看出, 吸附后的s分態密度曲線向低能量區移動約7.5 eV, 且能量為-15.5 eV ~ -14.5 eV處的態密度曲線僅由s分態密度貢獻. 吸附前CH4的s分態密度曲線峰值約為6.0 electrons/eV與7.0 electrons/eV, 而吸附后2個峰值分別約為4.0 electrons/eV、 3.4 electrons/eV, 分別降低約2.0 electrons/eV和4.4 electrons/eV, 表明吸附后CH4的s態能量降低, CH4分子的電子結構更加穩定.
圖6(c)為吸附前后CH4的p分態密度圖, 圖中天藍色曲線為吸附前CH4的p分態密度, 深藍色曲線為吸附后CH4的p分態密度. 吸附前, 能量主要分布在-0.75 eV ~ 0.75 eV, 吸附后的能量主要分布在-8 eV ~ -6.5 eV. 吸附前CH4態密度曲線峰值約為10.5 electrons/eV, 而吸附后峰值約為5.7 electrons/eV, 降低約4.8 electrons/eV, 表明吸附后p分態能量降低同時結構更加穩定. 綜上可得, 吸附作用對CH4分子的電子態密度的分布有著十分顯著的影響.
(3)吸附前后H2O的態密度
為了更好的分析H2O在SBi位吸附前后H2O的電子態密度變化, 將吸附前后H2O的總態密度、s態和p態分態密度繪制在同一圖形中進行了對比分析, 如圖7所示.
圖7(a)為H2O在SBi位吸附前后的總態密度圖, 圖中黑色曲線為吸附前H2O的總態密度, 紅色曲線為吸附后H2O的總態密度, 吸附后的態密度曲線整體向低能量區移動約5 eV, 吸附前, 能量主要分布在-18.7 eV ~ -17.5 eV、 -6.5 eV ~ -5.0 eV、 -2.7 eV ~ -1.3 eV、 -0.7 eV ~ 0.7 eV和高能區的5.7 eV ~ 17.0 eV, 吸附后, 能量主要分布在-21.0 eV ~ -19.7 eV、 -9.0 eV ~ -7.5 eV、 -5.0 eV ~ -3.8 eV、 -3.0 eV ~ -1.9 eV和3.0 eV ~ 14.0 eV. 吸附前H2O態密度曲線有4個約4.4 electrons/eV的峰值和1個約2.5 electrons/eV的峰值, 而吸附后4個4.4 electrons/eV的峰值降低為3.8 electrons/eV, 2.5 electrons/eV的峰值降為2個1.0 electrons/eV的峰值, 即吸附后H2O的總軌道數降低, 表明吸附后能量降低的同時結構更加穩定.
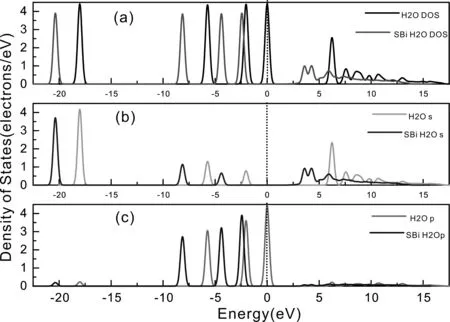
圖7 SBi位吸附前后H2O的態密度Fig.7 Densities of states of H2O before and after adsorption on SBi site
圖7(b)為吸附前后H2O的s分態密度圖, 圖中綠色曲線為吸附前H2O的s分態密度, 藍色曲線為吸附后H2O的s分態密度. 由圖可知, 吸附前-18.7 eV ~ -17.5 eV、 -6.4 eV ~ -5.0 eV和5.7 eV ~ 7.0 eV處的s分態密度曲線峰值分別由4.2 electrons/eV、 1.4 electrons/eV和2.4 electrons/eV降為吸附后-21.0 eV ~ -19.7 eV、 -9.0 eV ~ -7.5 eV和3.0 eV ~ 4.8 eV處的3.7 electrons/eV、 1.2 electrons/eV和2個1.0 electrons/eV. 由s分態密度圖與總態密度圖可知, 吸附前-18.7 eV ~ -17.5 eV、 5.7 eV ~7.0 eV與吸附后-21.0 eV ~ -19.7 eV、 3.0 eV ~ 4.8 eV處的總態密度主要由s分態貢獻, 即s分態占據高能量區與低能量區的電子軌道.
圖7(c)為吸附前后H2O的p分態密度圖, 圖中粉紅色曲線為吸附前H2O的p分態密度, 藍黑色曲線為吸附后H2O的p分態密度. 吸附前-6.5 eV ~ -5.0 eV、 -2.7 eV ~ -1.3 eV和-0.7 eV ~ 0.7 eV處的p分態密度曲線峰值分別由3.0 electrons/eV、 3.6 electrons/eV、 4.4 electrons/eV降為-8.9 eV ~ -7.5 eV、 -5.0 eV ~ -3.8 eV、 -3.0 eV ~ -1.9 eV處的2.7 electrons/eV、3.2 electrons/eV、3.8 electrons/eV. 由p分態密度圖與總態密度圖對比可知, 吸附前-6.5 eV ~ -5.0 eV、 -2.7 eV ~ -1.3 eV、 -0.7 eV ~ 0.7 eV與吸附后-8.9 eV ~ -7.5 eV、 -5.0 eV ~ -3.8 eV、 -3.0 eV ~ -1.9 eV處的總態密度主要由p分態貢獻, 即p分態占據費米能級下較高能量區的電子軌道.
5 結 論
基于密度泛函理論第一性原理方法, 研究了CH4與H2O在CaCO3(010) 面的吸附, 優化并計算了CH4與H2O在CaCO3(010) 面高對稱位的T位、 B位和H位的吸附模型結構, 計算了各高對稱位的吸附能, 并對CH4與H2O各自最穩定的吸附位吸附前后的物理結構和電子態密度進行了對比分析. 結果表明:
(1)CH4和H2O分子在CaCO3(010)面的吸附不明顯, 為物理吸附; CH4分子在LBⅢ位吸附能最小(-0.405 eV), H2O分子在SBⅰ位吸附能最小(-0.138 eV); CH4分子在CaCO3(010) 面的吸附性強于H2O分子, 宏觀表現為CaCO3親氣.
(2)對CH4與H2O各自最穩定吸附的位置吸附前后的物理結構進行分析發現, 鍵長鍵角的變化較小, 表明吸附作用對CH4和H2O分子結構的影響較小.
(3)LBⅢ位態密度曲線峰值整體較SBⅢ大, 能量范圍也較SBⅢ位態密度稍大, 故CH4在LBⅢ位更穩定; H2O在SBi位態密度曲線穩定性優于Tiii位, 即H2O在SBi位吸附更穩定.
(4)吸附后CH4的態密度曲線整體向低能量區偏移約7.5 eV,s分態、p分態能量降低,吸附后其結構更加穩定; 吸附后H2O的態密度曲線整體向低能量區移動約5 eV, 總軌道數降低, 表明吸附后能量降低的同時結構更加穩定;s分態占據高能量區與低能量區的電子軌道;p分態占據費米能級下較高能量區的電子軌道.
