Wilson病疑似自身免疫性肝炎1例及文獻復習
2019-05-08 03:13:58
國際消化病雜志 2019年2期
肝豆狀核變性由Wilson在1912年首先報道,故又稱為Wilson病,是由ATP7B基因突變引起銅在肝臟及其他組織中沉積,導致肝臟、神經和精神系統異常的一種常染色體隱性遺傳的罕見疾病,經Wilson病診斷積分≥4分可確診。臨床上,成人型Wilson病常難以與自身免疫性肝炎(AIH)鑒別,導致患者無法得到準確而及時的治療。現報道上海交通大學醫學院附屬仁濟醫院診治的1例Wilson病患者,并對此罕見疾病進行文獻復習。
1 病例資料
患者女性,30歲,因“發現肝功能反復異常1年半”于2017年9月18日入院。患者于2016年3月17日孕16周時發現肝功能異常:丙氨酸氨基轉移酶(ALT)113 U/L,天冬氨酸氨基轉移酶(AST)154 U/L,谷氨酸轉肽酶(GGT)48 U/L,堿性磷酸酶(ALP)107 U/L;乙型肝炎兩對半:HBsAg陽性,HBeAb陽性,HBcAb陽性;HBV-DNA<20 IU/mL,甲、丙、丁、戊型肝炎抗體均為陰性。EB病毒(EBV)、巨細胞病毒(CMV)抗體均為陰性。腹部B超未見明顯異常。外院診斷為“妊娠合并肝損、非活動性HBsAg攜帶者”,予復方甘草酸苷、腺苷蛋氨酸保肝治療,肝功能改善后出院(ALT 47 U/L,AST 72 U/L)。2016年9月14日孕38周順產一名男嬰。產后出現發熱,于外院檢查發現氨基轉移酶進行性升高:ALT、AST、GGT、ALP分別升至324 U/L、433 U/L、88 U/L、158 U/L;總膽汁酸(TBA)15.7~27.6 μmol/L;HBV-DNA<20 IU/mL,甲、丙、丁、戊型肝炎病毒標志物均陰性。EBV衣殼抗原IgA抗體陽性,EBV-DNA 8.910 7 copies/mL。腹部彩超未見明顯異常。遂診斷為“EBV感染伴肝損、非活動性HBsAg攜帶者”,予更昔洛韋抗病毒、復方甘草酸苷、腺苷蛋氨酸保肝治療,肝功能改善后出院(ALT 89 U/L,AST 109 U/L )。2017年9月10日外院復查腹部B超提示肝硬化,脾大。肝功能再次異常:ALT 125 U/L,AST 144 U/L,GGT 72 U/L,ALP 108 U/L。HBsAg陽性,HBeAb陽性,HBcAb陽性,HBV-DNA<20 IU/mL,甲、丙、丁、戊型肝炎病毒標志物均陽性。EBV衣殼抗原IgA抗體陽性,EBV-DNA陰性,CMV-Ab陰性,自身免疫性抗核抗體譜陰性,IgG 17.8 g/L,IgM 2.17 g/L。于2017年9月15日行肝穿刺活組織病理檢查,結果顯示:小葉結構紊亂伴假小葉形成,門管區、纖維間隔內中-重度慢性炎性反應伴中度界面性肝炎(圖1A),炎性細胞主要為淋巴細胞,可見漿細胞及中性粒細胞,部分肝細胞呈“玫瑰花環”樣排列(圖1B)。CK7免疫組化檢查示門管區、纖維間隔膽管明顯增生。銅染色可疑陽性。病理診斷為:慢性肝炎,肝硬化形成(G3,S4)。門診診斷為:(1)肝炎后肝硬化,自身免疫性肝炎?(2)非活動性HBsAg攜帶者。患者既往5年前婚檢時發現乙型肝炎小三陽、肝功能正常,腹部B超無異常;有輕度貧血史,未診治;有橋本甲狀腺炎病史;否認長期服藥史、否認吸煙飲酒史。2012年順產一足月男嬰;2014年因前置胎盤,孕6月時流產,這兩次孕期均無肝功能異常史。2016年孕38周時順產一男嬰,妊娠期有肝功能異常及TBA升高史。患者為獨生女,父母否認慢性肝炎史,叔叔因“肝硬化”于30歲時病逝。患者入本院后查體無明顯異常,繼續完善相關輔助檢查。自身免疫性肝病抗體:抗核抗體(ANA)1∶100,抗纖維肌動蛋白抗體(F-actin)35.19 IU,余抗體陰性。免疫球蛋白組合:IgG 17.2 g/L(7~16 g/L), IgM 1.61 g/L(0.4~2.3 g/L)。銅藍蛋白(CPN)0.09 g/L(0.2~0.6 g/L),血清銅8.9 μg/dL(11.8~39.3μg/dL),尿銅193.4 μg/24 h(0~60 μg/24 h)。顱腦MRI未見異常。基因檢測:ATP7B基因復合雜合突變(p.Thr935Met,p. Ser975Phe)。眼科會診未見Kayser-Fleischer(K-F)環。遂確診為:Wilson病,非活動性HBsAg攜帶者。予鋅劑口服治療(每日3次,每次50 mg),3個月后隨訪肝功能恢復正常。
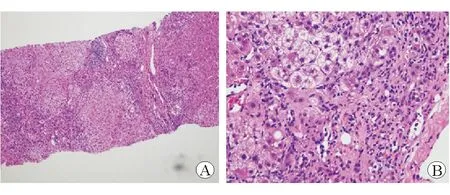
圖1肝臟活組織病理檢查 蘇木精-伊紅染色A×100B×200
2 討論
Wilson病是由細胞銅轉運缺陷,導致銅在肝臟及其他組織(包括腦)內蓄積,從而導致肝臟、神經系統和精神系統的異常表現[1]。該病分布于世界各地,人群中的患病率約為3萬分之一[2]。男性和女性的患病率大致相同,但女性較男性更易發生急性肝衰竭[3-5],而男性較女性更易發生神經精神性疾病[6]。Wilson病是一種常染色體隱性遺傳病,由于13號染色體上編碼銅轉運蛋白ATP7B的基因發生突變[7],導致銅與CPN的結合以及向膽汁中的排泄減少,從而在肝臟中蓄積。過量的肝銅使自由基產生增多,對細胞器的氧化損傷增加,導致肝細胞損傷。肝臟銅含量增加以及肝細胞損傷導致銅釋放入血,因此推測血清游離銅(不與血清CPN結合)增加是肝外銅沉積和隨后在腦及其他組織中產生毒性的原因。
大多數Wilson病患者在5歲至35歲之間被確診。Wilson病的臨床表現主要在肝臟、神經系統和精神疾病方面,許多患者具有這些癥狀的組合。肝臟表現包括伴有Coombs陰性溶血性貧血的急性肝功能衰竭、急性肝炎、慢性肝炎、肝硬化、肝脂肪變性和無癥狀的肝臟生化異常。神經系統表現廣泛,大多數神經性Wilson病的患者具有構音障礙和(或)運動障礙,依據此診斷該病具有一定挑戰性。其他臨床表現有Coombs陰性溶血性貧血和K-F環。Wilson病的輔助檢查中尿銅、CPN、K-F環的敏感度和特異度均不高。該病的肝臟活組織檢查也呈各種類型的病理改變,包括脂肪變、氣球樣變、Mallory小體、界面炎、橋接壞死、肝硬化,只有10%的患者肝銅染色為陽性。
Wilson病的診斷尚存在這些困難:(1)發病年齡懸殊(3歲~80歲);(2)多臟器累及;(3)初發癥狀多樣,臨床表現復雜多變(從輕微肝功能異常到暴發性肝衰竭);(4)輔助檢查的敏感度和特異度不高;(5)基因檢測尚未普及;(6)臨床醫生對該疾病的認識不足。因此,對于具有疑似Wilson病臨床特征的患者,應首先檢測肝功能、血常規、血清CPN和血清銅水平、眼裂隙燈檢查和24 h尿銅。這些檢查的結果決定了進一步行肝穿刺或組織病理檢查和基因檢測的必要性。目前,Wilson病的診斷依據2001年萊比錫國際會議制定的診斷評分系統[8]。本病例為中年女性,以反復肝功能異常為臨床表現,其中血清CPN<0.1 g/L,尿銅>2倍正常值上限(ULN),肝組織銅染色可疑陽性,基因檢測發現復合雜合子突變,可評8~9分(見表1),故確診為Wilson病。
自身免疫性肝炎(AIH)是一種慢性炎性肝病,女性多發,分布年齡廣,通常以自身抗體和血清球蛋白水平升高為特征,臨床表現變化大(從無癥狀到急性肝衰竭)。其診斷基于排除其他慢性肝病后的特征性血清學和組織學表現(包括界面性肝炎、匯管區和小葉內淋巴漿細胞浸潤和肝細胞玫瑰花結)。AIH初始治療通常采用糖皮質激素,大部分患者對激素應答良好。在早期的診治中,因本病例符合AIH的簡化診斷標準[9],評分7分可確診,故極易誤診為AIH;而通過AIH綜合診斷積分系統[10],評分14分,只能得出AIH可能的結論。患者后期鋅劑單藥治療后肝功能改善也說明了本例患者的病因是一元論而非二元論。
總之,Wilson病十分罕見,患者的肝臟活組織病理檢查及血清免疫學特點與典型AIH相似。鑒于本病例依據AIH簡化診斷標準無法鑒別Wilson病,因此臨床上應高度注意:(1)對3歲~80歲不明原因的肝損傷、肝硬化患者,應常規排查Wilson病;(2)診斷AIH前應除外Wilson病,特別是激素應答不良、合并Coombs陰性溶血性貧血或神經系統癥狀的患者;(3)推薦CPN、24 h尿銅和K-F環作為Wilson病檢查的首選指標,而肝組織病理檢查并非診斷的充分必要條件;(4)K-F環陰性、CPN正常均不能除外Wilson病,CPN降低也不能確診Wilson病;(5)Wilson病患者應終身治療,定期隨訪評估。

表1 患者的Wilson病診斷評分
注:≥4分為診斷確立;3分為診斷可能,需更多檢查;≤2分為診斷非常不可能;患者總得分為8~9分;a或腦MRI的典型異常或UWDRS評分;b如無定量肝銅可用
猜你喜歡
肝博士(2024年1期)2024-03-12 08:38:08
肝博士(2022年3期)2022-06-30 02:48:58
中老年保健(2021年3期)2021-08-22 06:50:04
昆明醫科大學學報(2021年1期)2021-02-07 01:06:36
現代臨床醫學(2021年1期)2021-01-26 00:56:02
中華養生保健(2020年4期)2020-11-16 01:31:40
中西醫結合肝病雜志(2020年2期)2020-10-27 02:18:50
中國衛生標準管理(2015年1期)2016-01-14 03:41:20
中國藥業(2014年12期)2014-06-06 02:17:26
疑難病雜志(2014年12期)2014-04-16 05:19:29
