橡膠籽油皂腳制備端環氧基脂肪酸酯的研究
2019-05-13 09:20:02陶云鳳和宇娟張金成高祎林楊曉琴
中國油脂 2019年5期
謝 東,陶云鳳,和宇娟,張金成,高祎林,楊曉琴
(西南林業大學 云南省高校林木生物技術重點實驗室,西南山地森林資源保育與利用教育部重點實驗室,昆明 650224)
植物油是來源廣泛、價格低廉、可再生的綠色生物質資源,尤其是非食用木本植物油的開發利用對于拓寬生物基化學品和生物基材料的原料來源范圍意義重大[1-6]。橡膠籽作為橡膠產業的副產品,每年因得不到合理的開發利用而被大量廢棄[7]。橡膠籽油具有較高的不飽和度,是生物柴油、環氧增塑膠劑、脂肪酸、多元醇、聚氨酯等生物基化學品和材料的很好原料[7-9]。但是橡膠籽大規模提油過程中,產生的脂肪酸導致橡膠籽油有較高的酸價(KOH)(>20 mg/g),需脫酸降低酸價才有利于深入加工利用[10-11]。常用堿煉法進行脫酸,脫酸過程會產生皂腳[12]。目前皂腳加工產品附加值不高,很少進行深度加工利用,這樣不僅污染環境,而且造成資源的極大浪費,因此綜合利用油脂皂腳制備高附加值產品一直備受關注[13-15]。環氧植物油脂及其衍生物是一類重要的生物基化學品,作為綠色增塑劑和穩定劑可直接添加到聚氯乙烯(polyvinyl chloride,PVC)中以替代石化基增塑劑,經環氧開環反應可合成用于制備生物基聚氨酯材料的多元醇,也可作為聚合單體發生環氧開環聚合反應制備生物基高分子材料[16-21]。然而,常見的環氧油脂及其衍生物的環氧鍵位于脂肪酸鏈中間,合成過程中易發生開環等副反應[4],進一步聚合時易產生交聯而導致聚合產物性能較差。因此,本文以橡膠籽油皂腳為原料合成含有端環氧基的脂肪酸酯,為油脂基高分子材料提供良好的聚合單體,同時實現廢棄皂腳資源化利用。
1 材料與方法
1.1 實驗材料
1.1.1 原料與試劑
橡膠籽油皂腳(干基,皂含量≥90%,中性油含量≤5%),油酸鈉(純度≥96%)、四丁基溴化銨、環氧氯丙烷、石油醚(沸程6090℃)、硅膠(200~300目)及其他試劑均為市售分析純,購自昆明盤龍華森實驗設備成套部。
1.1.2 儀器與設備
德國Bruker AV300 型核磁共振儀; 美國Thermo IS50 傅里葉變換紅外光譜儀。
1.2 實驗方法
1.2.1 油酸鈉為原料合成端環氧基油酸酯
為了更好地研究橡膠籽油皂腳與環氧氯丙烷的反應,首先以油酸鈉為模型物對反應過程進行研究,反應式如下:
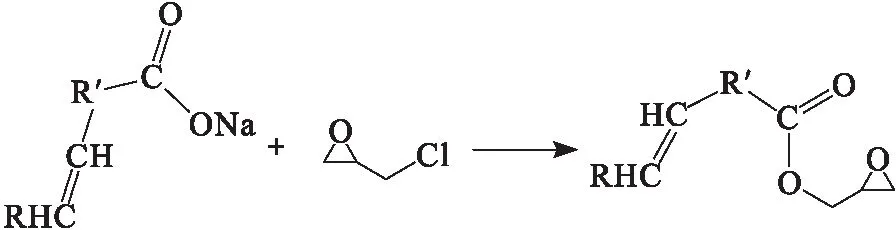
準確稱量 10 g 油酸鈉于150 mL三口燒瓶中,加入油酸鈉物質的量5%的四丁基溴化銨和一定量的環氧氯丙烷,攪拌下加熱反應一段時間,反應結束后冷卻至室溫,加入5 mL飽和碳酸氫鈉水溶液,用20 mL乙酸乙酯萃取3次,并依次用飽和氯化鈉水溶液、去離子水洗滌至中性,加入無水硫酸鎂干燥除水,過濾后旋轉蒸發除去溶劑即得端環氧基油酸酯粗品。
使用加壓硅膠柱層析純化端環氧基油酸酯粗品。將樣品用少量乙酸乙酯完全溶解后上柱,結合薄層色譜(TLC)分析收集比移值Rf為 0.5(UV)的組分,洗脫劑和展開劑均為石油醚-乙酸乙酯(體積比 25∶1),收集結束后濃縮除去溶劑,恒重即得純化的端環氧基油酸酯。得率按下式進行計算。
得率=實際端環氧基油酸酯質量/理論端環氧基油酸酯質量×100%
1.2.2 橡膠籽油皂腳為原料合成端環氧基脂肪酸酯
按1.2.1操作,將油酸鈉替換為橡膠籽油皂腳。
1.2.3 核磁分析
以TMS為內標,CDCl3作溶劑,在核磁共振儀上測定純化的端環氧基油酸酯的氫譜(1H NMR)和碳譜(13C NMR)。
1.2.4 FTIR分析
采用液體涂片法,掃描范圍400~4 000 cm-1。
2 結果與討論
2.1 反應條件的優化
以油酸鈉為模型物,探討了油酸鈉與環氧氯丙烷摩爾比、反應溫度和反應時間的影響。
2.1.1 油酸鈉與環氧氯丙烷摩爾比的影響
以四丁基溴化銨作相轉移催化劑,在反應溫度70℃、反應時間8 h條件下,研究了n(油酸鈉)∶n(環氧氯丙烷)對端環氧基油酸酯得率的影響,結果如圖1所示。
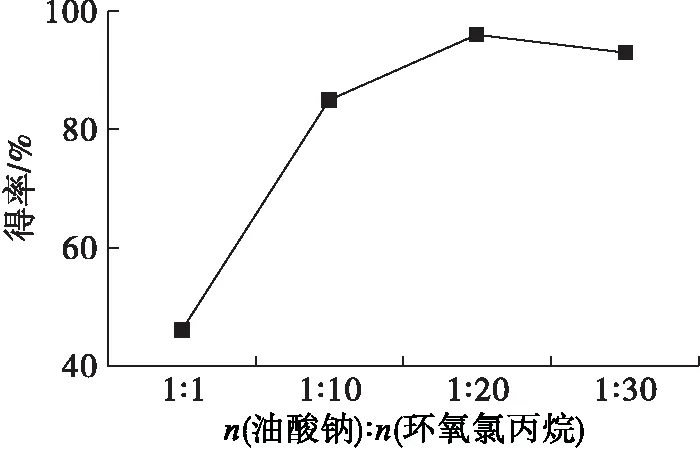
圖1 n(油酸鈉)∶n(環氧氯丙烷)的影響
由圖1可知,增加環氧氯丙烷的用量,產物的得率呈先增長后降低趨勢。在該反應中,環氧氯丙烷既是反應物,也充當了反應體系的溶劑,如果用量過少,反應物間的相互接觸面積小,反應速率慢。但過多的溶劑反而會稀釋反應體系,降低反應速率。因此,n(油酸鈉)∶n(環氧氯丙烷)為1∶20較佳。
2.1.2 反應溫度的影響
以四丁基溴化銨作相轉移催化劑,在n(油酸鈉)∶n(環氧氯丙烷)1∶20、反應時間8 h條件下,研究了反應溫度對端環氧基油酸酯得率的影響,結果如圖2所示。
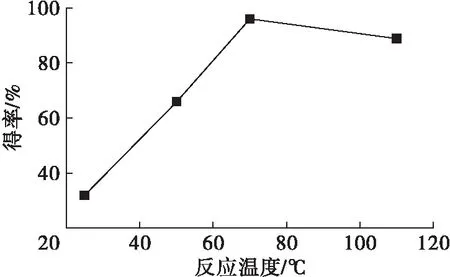
圖2 反應溫度的影響
由圖2可知,端環氧基油酸酯得率在一定范圍內隨著反應溫度的升高而增加,但過高的反應溫度會導致得率降低。由于環氧基是張力環,高溫下容易發生開環反應導致副產物增加,得率降低,不利于后期分離,但反應溫度過低,反應速率降低,因此實驗選擇在較溫和的反應溫度70℃下進行反應,既能減少副反應的發生,又保證反應效率。
2.1.3 反應時間的影響
以四丁基溴化銨作相轉移催化劑,在n(油酸鈉)∶n(環氧氯丙烷)1∶20、反應溫度70℃條件下,研究了反應時間對端環氧基油酸酯得率的影響,結果如圖3所示。
由圖3可知,端環氧基油酸酯得率隨著反應時間的延長先快速增加后緩慢降低,反應8 h 端環氧基油酸酯得率最高,但繼續延長反應時間不僅會導致產物進一步發生副反應,降低目標產物得率,且增加能耗。因此,反應時間8 h較佳。
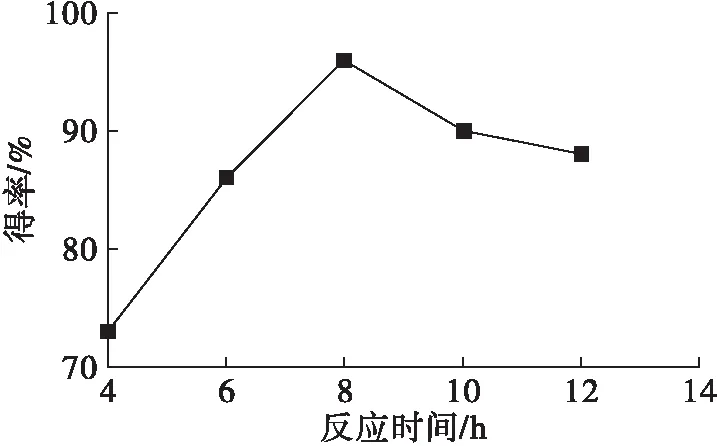
圖3 反應時間的影響
根據上述分析結果,確定合成端環氧基油酸酯的優化條件為:四丁基溴化銨作相轉移催化劑,n(油酸鈉)∶n(環氧氯丙烷)1∶20,反應溫度70℃,反應時間8 h。在優化條件下,端環氧基油酸酯得率較高,為96%。
2.2 結構表征
2.2.1 核磁分析
對優化條件下,以油酸鈉為原料合成的端環氧基油酸酯進行了1H NMR和13C NMR分析,結果如圖4和圖5所示。
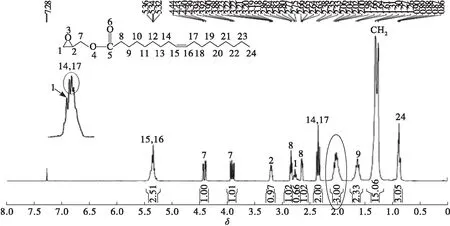
圖4 端環氧基油酸酯1H NMR 譜圖
由圖4可知,端環氧基油酸酯的1H NMR譜(300 MHz CDCl3δ)中δ2.77和δ2.06歸屬環氧基中亞甲基的兩個H—1,δ3.92歸屬環氧基中次甲基的一個H—2,說明環氧基是以端基的形式存在。δ4.39和δ3.90歸屬環氧基與酯基之間亞甲基的兩個H—7,δ2.64和δ2.84歸屬與羰基相連的亞甲基兩個H—8,δ1.64 歸屬羰基β位的亞甲基H—9,δ5.34歸屬烯氫中H—15和H—16,δ2.35是與烯鍵相連的亞甲基H—14和H—17特征信號,表明雙鍵的存在。δ1.26~δ1.31歸屬其余亞甲基中的H(10~13和18~23)特征信號,δ0.88歸屬端甲基H—24。
由圖5可知,端環氧基油酸酯的13C NMR譜(75 MHz CDCl3δ)中δ44.64和δ49.38分別歸屬環氧基C—1和C—2,δ64.75歸屬環氧基與酯基之間C—7,δ173.49歸屬酯基C—5,δ130.01歸屬烯碳C—15和C—16,δ14.10歸屬端甲基C—24,δ22.57~δ34.06 歸屬其余亞甲基C(8~14和17~23)。
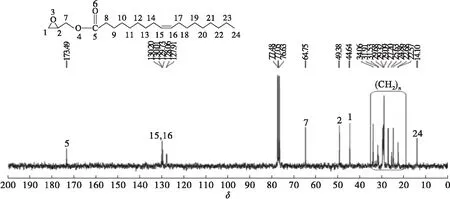
圖5 端環氧基油酸酯13C NMR 譜圖
2.2.2 FTIR分析
將2.1中優化的條件用于橡膠籽油皂腳制備端環氧基脂肪酸酯的反應中,產品得率為80%。對橡膠籽油皂腳制備的端環氧基脂肪酸酯產品進行FTIR分析,結果如圖6所示。
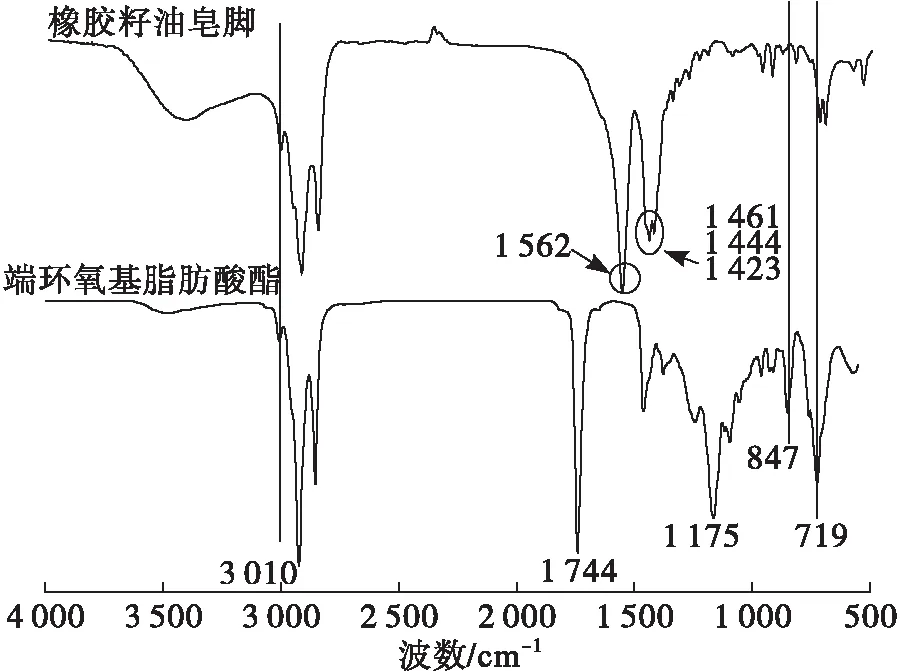
圖6 橡膠籽油皂腳和端環氧基脂肪酸酯的FTIR譜圖
由圖6可知,與橡膠籽油皂腳的譜圖相比,產品譜圖中3 010 cm-1中的雙鍵吸收峰未消失,皂腳的特征吸收峰1 562、1 461、1 444、1 423 cm-1消失,這些吸收峰是由于羧酸鹽 —COOM的羧基 —COO— 伸縮振動引起,經過反應,羧酸鹽轉化為酯,并在近羧基的碳鏈上引入了吸電子基團,使特征吸收移向高波數,轉而出現酯基在1 744 cm-1處的特征吸收峰。同時,產物中出現了847 cm-1和719 cm-1的環氧基特征吸收峰、1 175 cm-1的醚鍵C—O—C吸收峰,表明產物中脂肪酸鹽的消失和環氧鍵的形成。橡膠籽油皂腳制備的端環氧基脂肪酸酯雖為混合物,但端環氧基具有較高的反應活性,且保留了原結構中的雙鍵,提供了更多的改性空間。
3 結 論
以油酸鈉為模型物與環氧氯丙烷反應制備端環氧基油酸酯,通過單因素實驗得到優化的工藝條件為:四丁基溴化銨作相轉移催化劑,n(油酸鈉)∶n(環氧氯丙烷)1∶20,反應溫度70℃,反應時間8 h。在優化工藝條件下,端環氧基油酸酯得率為 96%。采用1H NMR和13C NMR對端環氧基油酸酯進行結構分析,表明了端環氧基被成功引入。
以優化的工藝條件用于橡膠籽油皂腳與環氧氯丙烷的反應中,以制備端環氧基脂肪酸酯,端環氧基脂肪酸酯的得率為80%。通過FTIR對產物進行分析,結果表明產物的結構中不僅引入了端環氧基,而且保留了雙鍵,為其進一步化學改性提供了更多的空間。
