熱處理時間對CuZnAl催化劑合成C2+OH性能的影響
2019-06-03 02:32:12鄧皓月高志華
燃料化學學報 2019年5期
關鍵詞:催化劑
鄧皓月, 高志華, 黃 偉
(太原理工大學 煤科學與技術教育部與山西省重點實驗室, 山西 太原 030024)
隨著近年來全球能源和環境壓力的增大,世界各國都加大了對非石油路線制取液體燃料的研究[1,2]。低碳醇(C2+OH)具有較高的辛烷值,不僅可以作為一種燃料添加劑,更可以直接用來替代傳統液體燃料。而中國“富煤、貧油、少氣”的能源結構決定了由煤為原料經合成氣制備C2+OH研究具有重要的工業應用價值[3-5]。
在合成氣制C2+OH反應過程中,高效催化劑的研發是該技術的關鍵所在。當前,煤基合成氣直接制備C2+OH所用的催化劑主要分為四類[6-8]:銠基催化劑、鉬基催化劑、改性F-T催化劑和改性甲醇催化劑。其中,銠基催化劑因其獨特的對C2+含氧化合物選擇性,被認為是合成氣制C2+OH的最佳催化劑。但貴金屬銠儲量太少,成本較高,難以工業化;而在非貴金屬催化劑中,鉬基催化劑雖然有著較好的抗硫性能,但總體活性不高,反應條件較為苛刻;改性F-T催化劑的F-T組元(Fe、Co、Ni)能增長碳鏈,提高C2+OH選擇性,但生成的產物種類較多,分離困難,穩定性較差;改性甲醇催化劑主要產物還是甲醇,C2+OH產量較低。因此,有必要研究制備高活性和高選擇性合成C2+OH的新型廉價催化劑,以便實現合成氣制C2+OH技術在工業上的規模化應用。
本課題組針對漿態床的使用特性,提出了一種全新的催化劑制備方法——完全液相法[9]。其創新之處在于催化劑的熱處理過程是在漿態床使用的惰性介質中完成的,避免了傳統方法制備環境與使用環境不一致導致催化劑分布不均甚至失活的問題。本課題組在前期工作中發現[10],采用完全液相法制備的不含F-T組元或堿金屬的CuZnAl催化劑對C2+OH表現出了較好的選擇性。但該催化劑的性能對結構非常敏感,導致催化劑的重復性較差,目前,研究尚未澄清催化劑結構與反應性能間的內在關聯。喻仕瑞[11]和劉勇軍[12]研究均發現,CuZnAl催化劑合成C2+OH需要Cu+-Cu0雙活性位。其中,Cu+有助于促進CHxCO-中間體的形成,而Cu0主要是吸附并活化CO分子。只有當Cu+-Cu0的協同作用達到平衡時,才能促進C2+OH的生成。董偉兵[13]發現,催化劑在高壓熱處理條件下內部物種數量發生了改變,并且生成了常壓下沒有出現的尖晶石,活性評價結果顯示乙醇選擇性增加明顯,不足之處是催化劑反應活性很低,而且沒有對高壓熱處理條件的影響進行更為深入的分析。因此,為了進一步研究高壓下熱處理條件對催化劑表面活性物種的影響,強化Cu+-Cu0的協同作用,本實驗在課題組前期研究成果上,采用完全液相法制備CuZnAl催化劑,考察高壓條件下不同熱處理時間對催化劑結構和性能的影響,為后續開發性能優異的催化劑提供借鑒和指導。
1 實驗部分
1.1 催化劑的制備
本實驗所用試劑如未特殊說明均來自天津市科密歐化學試劑有限公司。
催化劑組分物質的量比為Cu∶Zn∶Al=2∶1∶4。制備過程首先將40.9 g研磨好的異丙醇鋁(AR,天津市光復精細化工研究所)溶于70 mL乙醇中,75 ℃下攪拌2 h后在室溫下放置1 d。將溶液升溫至75 ℃,然后將含有24.2 g硝酸銅、14.9 g硝酸鋅、1 g聚乙二醇600(AR,上海市阿拉丁生化科技股份有限公司)和35 mL乙醇的混合溶液用蠕動泵緩慢滴加到上述溶液中,升溫至85 ℃,加入180 mL蒸餾水水解1 h。最后加入少量濃硝酸,升溫至95 ℃,回流攪拌直至形成凝膠。
將制得的凝膠在25 ℃下老化10 d后分散在250 mL液體石蠟中,置于500 mL高壓反應釜內進行熱處理。熱處理過程:N2流動氣氛下,壓力保持在1.0 MPa,以1 ℃/min的升溫速率從室溫升至280 ℃,恒溫熱處理一定時間以獲得不同的漿狀催化劑。熱處理時間為3.5、7、10.5 h的催化劑分別命名為Cat-3.5、Cat-7和Cat-10.5。
1.2 催化劑的活性評價
將熱處理后的催化劑置于500 mL高壓反應釜內進行活性評價。固含率為10%,不進行預還原。合成氣配比H2/CO(體積比) =2∶1,總流量為150 mL/min。反應器壓力維持在4.0 MPa,反應溫度為280 ℃,持續反應6 d。反應產物使用海欣GC-950氣相色譜儀檢測。氣相產物中的H2、CO和CO2用TCD檢測器檢測;烴類、未冷凝的醇類以及二甲醚用FID檢測器檢測。每24 h收集一次液相產物后再通過FID檢測器檢測分析。所有活性數據均在反應24 h催化劑性能穩定后測定。CO轉化率(xCO)和產物的選擇性(si)分別根據以下公式計算:
(1)
(2)
式中,i包括氣液相產物中除去CO外的所有含碳物質。
1.3 催化劑的表征
將漿狀催化劑用石油醚抽提5 d,以除去殘留的液體石蠟。抽提后,自然晾干得到固體催化劑后再進行表征。
XPS使用美國賽默飛公司的ESCALAB 250型X光電子能譜儀測定。真空度為7×10-8Pa,激發源為AlKα(hv= 1486.6 eV, 150 W),以表面污染碳C 1s峰(284.6 eV)作荷電效應校正。
XRD使用日本理學公司的D/MAX-2500型衍射儀測定。輻射源為CuKα,5°-85°掃描,掃描速率8(°)/min。
H2-TPR使用天津先權儀器廠的TP-5080多用吸附儀測定。將50 mg樣品在150 ℃下用He吹掃30 min,自然降溫至50 ℃。再以5% H2/N2混合氣(30 mL/min)為還原氣,10 ℃/min的速率升溫至500 ℃。用熱導檢測器檢測耗氫量。
NH3-TPD-MS使用天津先權儀器廠的TP-5080多用吸附儀測定。將100 mg樣品在280 ℃下用He吹掃30 min后降溫至50 ℃,然后吸附純氨30 min至飽和。再通He吹掃30 min以除去物理吸附的氨。以10 ℃/min的速率升溫至800 ℃。氨脫附曲線使用QIC-20型質譜檢測器檢測。
織構性質使用美國康塔公司的QDS-30型物理吸附儀測定。首先將樣品在200 ℃下脫氣5 h,然后在液氮中采用N2吸附法測定。比表面積通過BET方法計算,平均孔徑和孔容通過BJH方法計算。
2 結果與討論
2.1 催化劑的活性評價
圖1為CuZnAl催化劑CO加氫制C2+OH過程中活性評價數據隨反應時間的變化。其中,圖1中各點均為評價第2-6 d催化劑性能穩定后每天活性數據的平均值。由圖1可知,在熱處理時間為3.5 h時,活性隨反應時間呈現下降趨勢,在反應第5 d后保持相對穩定。而熱處理時間較長的兩種催化劑CO轉化率總體保持穩定。三種催化劑C2+OH在總醇中的占比總體上均隨反應時間的延長而下降,在反應到第5 d后保持穩定。通過這兩條變化曲線可以看到,所有催化劑的活性在反應前期總體上均隨反應時間的延長而下降,但在反應5 d后維持在一個穩定數值。對5 d活性數據進行整體平均,計算得到Cat-3.5、Cat-7和Cat-10.5三種催化劑CO轉化率分別為40.9%、38.1%和37.8%,總醇(ROH)選擇性分別為21.8%、15.8%和13.0%,均隨著熱處理時間的延長而下降。而C2+OH在總醇中的質量分數分別為37.8%、65.9%和44.0%。總體來看,Cat-7催化劑性能最佳。
圖2為不同熱處理時間下催化劑合成C2+OH的產物碳數分布。
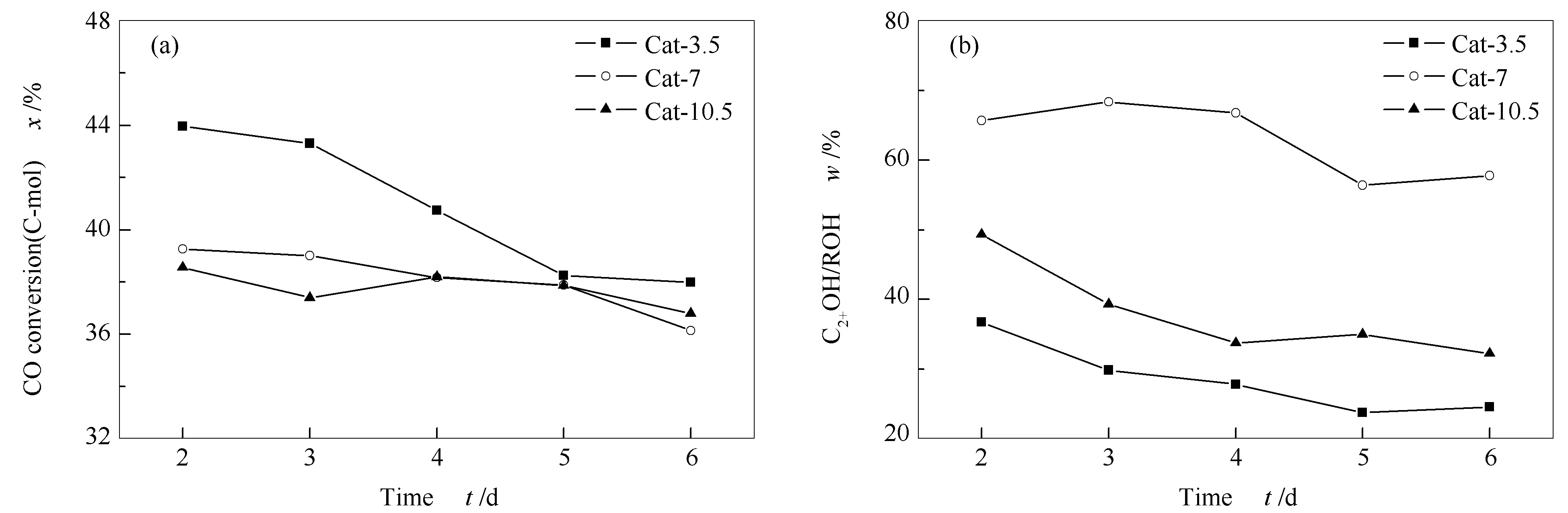
圖 1 CuZnAl催化劑在不同熱處理時間下的催化性能
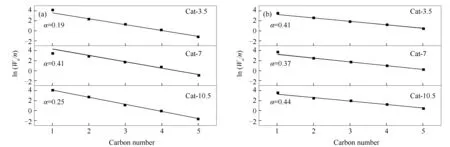
圖 2 CuZnAl催化劑在不同熱處理時間下產物的碳數分布
由圖2可知,所有催化劑的醇類和烴類產物均較好地服從ASF分布[14]。在醇產物分布中,Cat-3.5催化劑和Cat-10.5催化劑的鏈增長因子(α)僅為0.19和0.25,而Cat-7催化劑的鏈增長因子達到了0.41,這與Cat-7催化劑中C2+OH占比最高相對應。在烴產物分布中,所有催化劑的鏈增長因子均在0.40附近。催化劑表面醇、烴產物分布的鏈增長因子變化趨勢存在明顯差異,表明反應產物中的醇和烴是在催化劑不同活性位點上生成的[15,16]。由此可見,改變熱處理時間主要改變了醇類產物的碳鏈增長能力,選擇適宜的熱處理時間更容易形成長鏈的醇類產物。
2.2 XPS表征
圖3為催化劑反應前的Cu 2pXPS譜圖。由圖3可知,所有催化劑均沒有在940-945 eV間檢測到明顯的shake-up峰,表明催化劑表面沒有CuO物種[17]。各催化劑的Cu 2p3/2峰型均不對稱,經分峰擬合后存在兩個峰。其中,結合能位于932 eV附近的峰表明催化劑中Cu物種是以Cu+和Cu0形式存在[18-20];而結合能位于934.5 eV附近的峰則歸屬于尖晶石(CuAl2O4)的峰[20, 21]。尖晶石峰面積大小次序為Cat-10.5> Cat-7> Cat-3.5,這表明,催化劑在高壓熱處理過程中可以生成尖晶石物相,并且該物相數量是隨著熱處理時間的延長而增多的。文獻報道[22]催化劑生成尖晶石后活性位會減少,從而導致催化劑的活性下降,這與催化劑活性評價結果相一致。此外,XPS數據中所有催化劑的Zn 2p3/2結合能均在1021.5 eV附近,對應的Zn LMM譜峰在988 eV附近,表明Zn物種在催化劑中均以ZnO形式存在[23]。可見改變高壓熱處理的時間并沒有對Zn的存在形態產生影響。
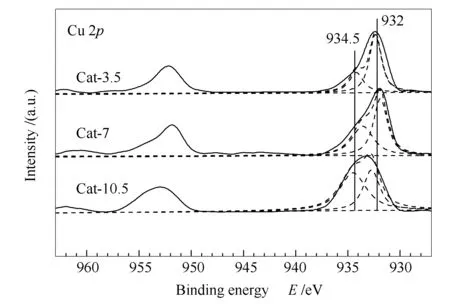
圖 3 CuZnAl催化劑在不同熱處理時間下的Cu 2pXPS譜圖
表1為催化劑表面各物種的元素含量。由表1可知,催化劑表面Cu/Zn和Cu/Al均小于催化劑的投料物質的量比(Cu/Zn/Al=2∶1∶4),這是因為Zn和Al的晶格能大于Cu,更容易在催化劑表面富集。Cat-7催化劑表面Cu/Zn最大,而高的Cu/Zn有利于C2+OH的生成[24]。隨著熱處理時間的延長,催化劑表面C含量下降明顯,說明延長熱處理時間能有效抑制液體石蠟在催化劑表面的堆積。
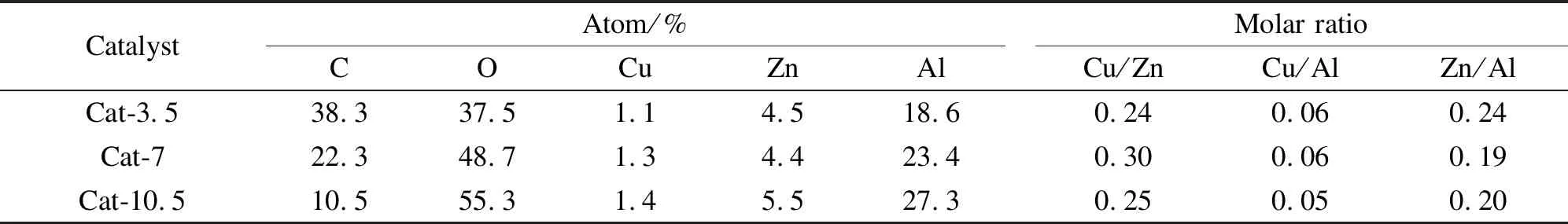
表 1 CuZnAl催化劑在不同熱處理時間下的表面元素組成
2.3 XRD表征
圖4為催化劑反應前的XRD譜圖。由圖4可知,所有催化劑均檢測到了Cu0的衍射峰。Cat-3.5催化劑中出現了AlOOH和ZnO的衍射峰,沒有檢測到尖晶石(CuAl2O4)的衍射峰。結合XPS結果,這是由于Cat-3.5催化劑中尖晶石物相數量較少,未達到XRD檢測線。Cat-7催化劑和Cat-10.5催化劑中均檢測到了尖晶石相的衍射峰,但沒有出現AlOOH衍射峰,可見熱處理時間的延長使催化劑中Cu、Al物種間的結合力增加,形成了更完整的尖晶石結構,從而一方面導致AlOOH數量減少;另一方面,也提高了AlOOH在體系中的分散度。同時從圖4可見,Cat-7和Cat-10.5催化劑中也未檢測到ZnO的衍射峰。本課題組前期的實驗結果表明[13],尖晶石的存在會使ZnO晶粒變小,分散度增加。本工作實驗結果進一步驗證了尖晶石的存在有助于提高AlOOH和ZnO的分散度。由謝樂公式計算出三種催化劑的Cu0晶粒粒徑分別為24.0、29.5和30.5 nm。這表明隨著熱處理時間的延長,催化劑中Cu0晶粒逐漸增大,晶型更為完整。文獻報道[25-27],Cu0晶粒越小,分散度越大,其對CO分子的吸附活化能力越強。而在CO加氫生成醇類和烴類過程中,催化劑表面存在較高的CO濃度可以促進CHx中間體插入CO生成醇類的前驅體CHxCO-,從而有利于醇類的生成。可見,Cu0晶粒越小,越有利于醇類產物的生成。在活性數據中,總醇選擇性隨熱處理時間的延長而下降,這與上述分析一致。
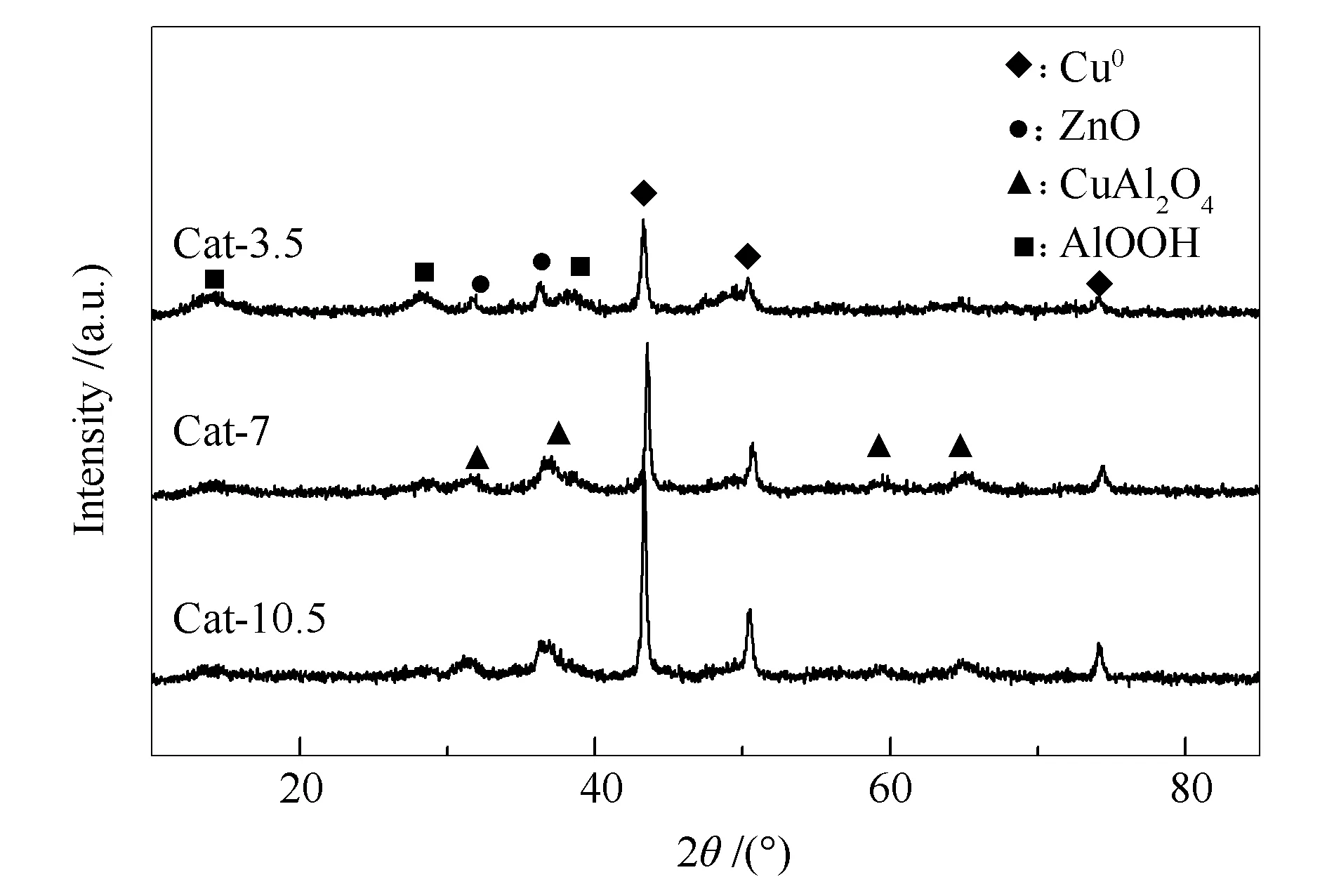
圖 4 CuZnAl催化劑在不同熱處理時間下的XRD譜圖
2.4 H2-TPR表征
圖5為催化劑反應前后的H2-TPR譜圖。由圖5可知,反應前后所有催化劑均出現了兩個還原峰。根據前期研究[24]和XPS結果,溫度較低的還原峰歸屬為此催化劑中晶粒粒徑較小Cu+物種的還原;溫度較高的還原峰歸屬為此催化劑中晶粒粒徑較大Cu+物種的還原。反應前后,不同催化劑各還原峰的中心溫度與峰面積也不相同。Cat-7催化劑反應前的高溫還原峰中心溫度較高,峰面積最大,在反應后下降幅度不大,表明合適的熱處理時間會使催化劑中存在較多晶粒粒徑大的,在反應過程中相對難被還原的Cu+。需要指出的是,Cat-10.5催化劑反應后的還原峰面積變大,這可能是由于反應過程中水煤氣變換反應產生的CO2、H2O等氧化物使Cu0氧化形成Cu+,類似現象在合成甲醇的文獻中也有報道[28]。但是僅有Cat-10.5催化劑出現此現象的原因目前還沒有明確結論。
前期采用完全液相法制備催化劑時,由于在常壓熱處理過程中液體石蠟受熱分解產物會將大部分Cu氧化物還原為Cu0,導致催化劑中Cu+較少[29]。研究發現,CuZnAl催化劑中Cu+-Cu0是合成C2+OH催化劑的雙活性中心。其中,Cu+有助于促進CHxCO-中間體的形成,而Cu0主要起到吸附活化CO分子的作用[11,12]。當催化劑中存在適量比例的Cu+和Cu0時,有利于Cu+-Cu0的協同作用的發揮,從而促進C2+OH的生成。本研究中高壓下熱處理適宜時間(7 h),有利于催化劑中形成較多難被還原的Cu+,使Cu+和Cu0的量達到平衡,進而提高了C2+OH在總醇中的占比。
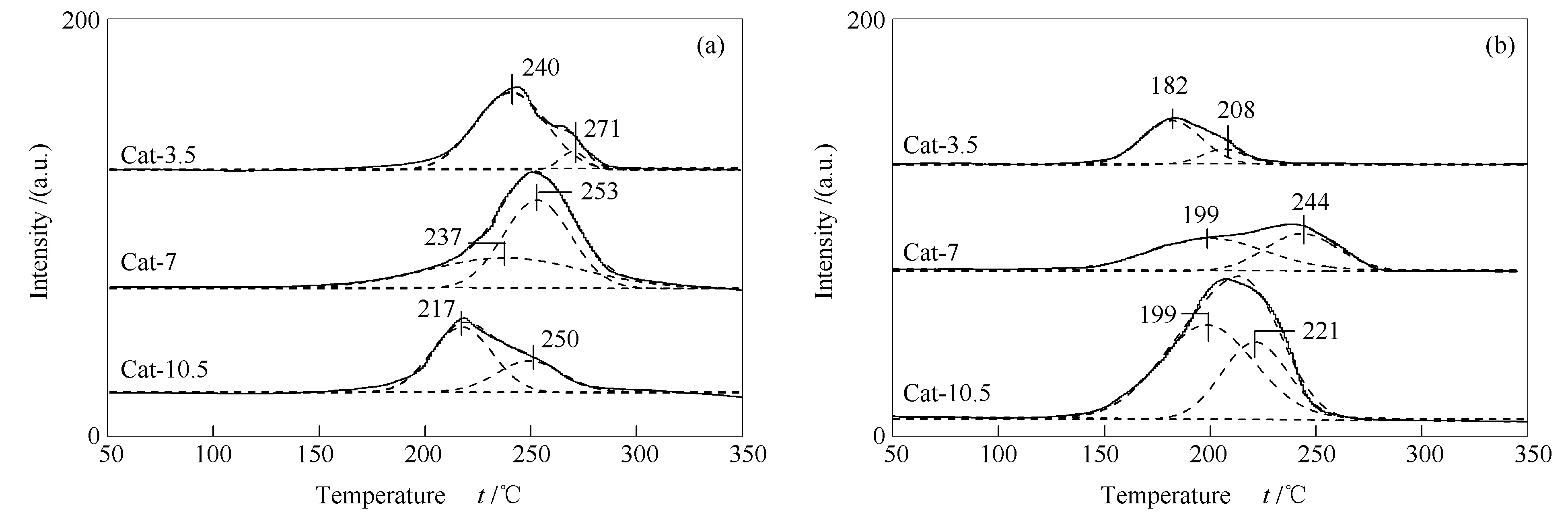
圖 5 CuZnAl催化劑在不同熱處理時間下的H2-TPR譜圖
2.5 NH3-TPD表征
圖6為催化劑反應前的NH3-TPD-MS譜圖。由圖6可知,所有催化劑均檢測到了兩個NH3脫附峰。其中,在150 ℃附近的脫附峰對應于催化劑表面的弱酸位;在550 ℃附近的脫附峰對應于催化劑表面的中強酸位。隨著熱處理時間的延長,催化劑表面弱酸位和中強酸位脫附峰面積均減小,表明延長熱處理時間會減小催化劑的表面弱酸量與中強酸量。通過對不同酸性位的脫附峰面積進行積分計算,三種催化劑表面弱酸量在總酸量中的占比分別為0.33、0.27和0.32,Cat-7催化劑的表面弱酸量占比最小。Liu等[23]認為,催化劑中較多的弱酸量有利于C2+OH的生成。呂曉東[30]認為,表面弱酸位更有利于二甲醚的生成。而董偉兵[13]卻發現,適當的弱酸量與中強酸量比例才有利于C2+OH的生成。可見,雖然各研究者在制備C2+OH時都選擇CuZnAl催化劑,但由于制備方法、處理條件等的差異,使得結果不盡相同。而本研究發現催化劑中較小的弱酸占比有利于C2+OH的生成。
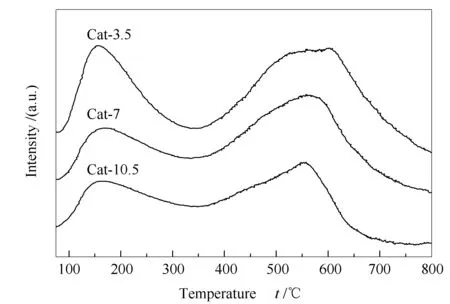
圖 6 CuZnAl催化劑在不同熱處理時間下的NH3-TPD-MS譜圖
2.6 織構參數
表2為催化劑反應前后的織構參數。反應前,隨著熱處理時間的延長,催化劑的比表面積逐漸減小,孔容和孔徑逐漸增大。經分析,高溫高壓條件下較長的熱處理時間會使催化劑中活性組分團聚,晶粒粒徑增大,從而導致比表面積減小;而孔容和孔徑逐漸增大是因為長的熱處理時間使催化劑孔道中的液體石蠟在N2流動氣氛下更多地被帶出,形成了更大的孔容和孔徑。相比其他兩種催化劑,Cat-3.5催化劑反應后的比表面積和孔容增大,這是由于其熱處理時間較短,催化劑孔道中殘留有較多液體石蠟,經過反應過程,殘留的液體石蠟逐漸解吸導致催化劑比表面積和孔容增大[31,32],這與XPS結果中Cat-3.5催化劑的表面碳含量最大相對應。Cat-7和Cat-10.5催化劑反應后比表面積和孔容均有所減小則可能是催化劑孔道在反應過程中被長鏈烴等產物堵塞所致。一般來說,大的孔容能為碳鏈增長提供足夠的空間,從而生成長碳鏈產物[33]。而較大孔徑有利于H2和CO在催化劑孔道中擴散,形成限域效應促進C2+OH的生成[34]。
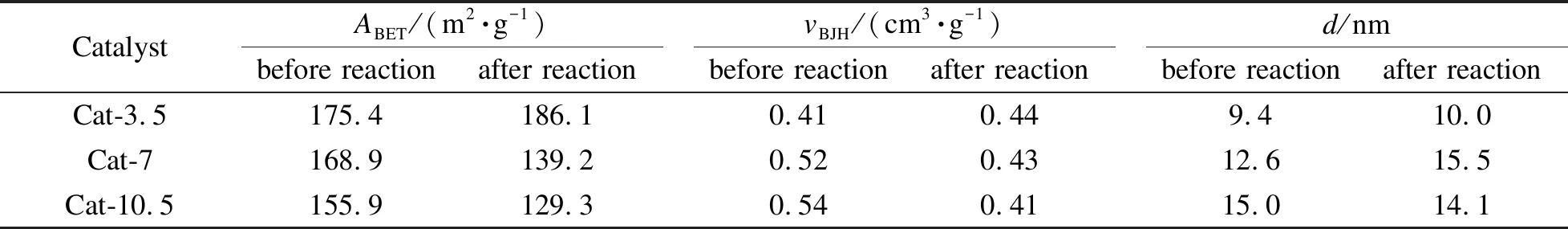
表 2 CuZnAl催化劑在不同熱處理時間下的織構參數
notes:ABET: BET surface area;vBJH: pore volume;d: average pore diameter
3 結 論
采用完全液相法制得的CuZnAl催化劑,不同熱處理時間主要對催化劑在CO加氫合成C2+OH反應過程中的醇類產物生成產生一定影響。通過表征結果發現,熱處理時間為7 h時,催化劑中存在較多難被還原的Cu+,此時Cu+-Cu0活性位點間的協同作用較好,C2+OH在總醇中的占比最高。而隨著熱處理時間的延長,催化劑中Cu、Al物種之間結合力增強,尖晶石的量隨之增多, CO轉化率下降。同時,熱處理時間的延長也使催化劑表面酸量減少,孔容和孔徑增大,而催化劑表面較小的弱酸占比、大的孔容和孔徑均有利于C2+OH的生成。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
