阻斷輔因子NADPH合成對谷氨酸棒桿菌生長及產物合成的影響
2019-06-06 06:51:50楊漢昆徐建中張偉國
食品與發酵工業 2019年10期
楊漢昆,徐建中*,張偉國*
1(江南大學 生物工程學院,江蘇 無錫,214122) 2(工業生物技術教育部重點實驗室(江南大學),江蘇 無錫,214122)
還原型煙酰胺腺嘌呤二核苷酸磷酸(nicotinamide adenine dinucleotide phosphate, NADPH)又稱為還原型輔酶Ⅱ,在很多生物體內的化學反應中起遞氫體的作用,具有重要的生物學意義。NADPH在細胞內分布廣泛,通過參與800多個氧化還原反應來調節細胞內氧化還原水平并影響著眾多基因表達、細胞功能、代謝途徑和物質跨膜運輸[1],參與多種合成代謝反應,如氨基酸、脂類及核苷酸等細胞組成物質的合成均需要NADPH提供還原力,對細胞正常生長和代謝有重要影響[2],并且是微生物代謝網絡中含量最豐富的氧化還原輔酶之一。胞內NADPH的生成和消耗同胞內很多重要的代謝途徑相關聯,維持細胞內輔酶的平衡對于細胞生長、代謝以及產物的合成都非常關鍵。煙酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide, NAD)是另一種極為重要的核苷酸類輔酶,是代謝網絡中的關鍵輔因子之一,在胞內參與超過300個氧化還原反應[3]。如在糖酵解、糖異生、三羧酸循環以及呼吸鏈等代謝中發揮著不可替代的作用[4-6]。NADH/NAD+這對輔因子在葡萄糖分解代謝中起著核心作用,以NAD+作為輔因子將葡萄糖氧化,并且NAD+同時轉化成等量的NADH還原形式。
NADPH對谷氨酸棒桿菌(C.glutamicum)合成L-賴氨酸非常重要,根據C.glutamicumL-賴氨酸合成代謝網絡可知有4個反應涉及NADPH的消耗,即合成1 molL-賴氨酸需要消耗4 mol NADPH[7]。研究結果表明在谷氨酸棒桿菌中NADPH的供應與消耗是不平衡的。在谷氨酸棒桿菌中分別由葡萄糖-6-磷酸脫氫酶、6-磷酸葡萄糖酸脫氫酶、蘋果酸酶和異檸檬酸脫氫酶以NADP+為輔因子,參與NADPH合成[8-9]。谷氨酸棒桿菌中的NADPH不僅要滿足L-賴氨酸合成需求而且還要用于菌體生長。因為,增加1g菌體需要消耗16.4 mmol NADPH[10]。目前的研究主要集中于通過輔因子工程提高NADPH的供應可以強化L-賴氨酸合成,而對阻斷胞內NADPH合成途徑對菌體生長及產物合成的影響研究匱乏。胞內NADPH水平可以改變胞內微環境,CHEN等[11]指出,改變胞內NADPH水平會擾動胞內的氧化還原水平和ATP含量;同時也會影響目標代謝產物產量,XU等[12]通過在谷氨酸棒桿菌中異源表達大腸桿菌膜結合轉氫酶PntAB來改變胞內NADPH水平,能夠影響谷氨酸棒桿菌合成L-賴氨酸的能力。氧化還原輔因子代謝工程成為優化生物轉化的重要代謝工程策略[13]。在谷氨酸棒桿菌中,研究表明L-賴氨酸的合成與輔因子NADPH水平密切相關[1]。
本研究以實驗室保存的L-賴氨酸生產菌株C.glutamicumLYS為出發菌株,敲除參與胞內NADPH合成的葡萄糖-6-磷酸脫氫酶、蘋果酸酶編碼基因,并將自身NADP+-依賴型異檸檬酸脫氫酶基因(icdCg)替換成變形鏈球菌(Streptococcusmutans)NAD+-依賴型異檸檬酸脫氫酶基因(icdSm)。對出發菌C.glutamicumLYS和重組菌C.glutamicumLYSΔzwfΔmalEΔicdCg::icdSm進行搖瓶實驗,分析阻斷谷氨酸棒桿菌中NADPH合成途徑對胞內NAD+、NADH、ATP、ADP和AMP、細胞生長、葡萄糖消耗速率、L-賴氨酸、有機酸以及其他副產物氨基酸合成的影響。為進一步研究NADPH調控L-賴氨酸產生菌胞內微環境的生理機制,調控和優化胞內NADPH與中心碳代謝的特異性,為改進微生物L-賴氨酸生產性能提供了研究基礎。
1 材料與方法
1.1 材料與試劑
1.1.1 菌株和質粒
C.glutamicumLYS、E.coliJM109,穿梭表達質粒pDXW-8和敲除質粒pK18mobsacB均為本研究室保藏,其他質粒均為實驗中構建。
1.1.2 主要試劑和儀器
限制性內切酶BamH Ⅰ、XbaⅠ、Hind Ⅲ、SalⅠ、EcoR Ⅰ,T4DNA連接酶、Taq DNA聚合酶、DNA Marker:購于寶生物工程有限公司;PCR相關試劑、基因組DNA提取試劑盒、DNA凝膠回收試劑盒、PCR產物純化試劑盒、質粒小量提取試劑盒:購于上海生工生物工程有限公司;輔酶Ⅱ NADP(H)和NAD(H)含量檢測試劑盒:購自北京索萊寶科技有限公司;其他試劑均為進口或國產分析純試劑。
UV 2100可見紫外分光光度計,尤尼柯(上海)儀器有限公司;JY 1600C凝膠水平電泳儀,北京六一儀器廠;SBA40-E生物傳感儀,山東省科學院生物研究所;G1-14高速離心機,Sigma公司;Gel DOC GR+凝膠成像系統,美國Bio-Rad公司;LC 1100高效液相色譜系統,安捷倫科技有限公司;Gene Pulser Xcell電穿孔儀,美國Bio-Rad公司。
1.1.3 培養基
LB培養基(g/L):蛋白胨 10,酵母粉 5,NaCl 10,pH 7.0。
LBG培養基(g/L):蛋白胨 10,酵母粉 5,NaCl 10,葡萄糖 5,pH 7.0。
Epo培養基(g/L):葡萄糖 5, 蛋白胨 10, 酵母膏 5, NaCl 10, 吐溫-80 1, pH 7.0,121 ℃滅菌20 min, 滅菌后加入無菌的異煙肼4 g和甘氨酸30 g,用于制備C.glutamicum感受態細胞。
LBHIS培養基(g/L):蛋白胨 5,酵母膏 2.5,NaCl 10,腦心浸出液 18.5,山梨醇 91,pH 7.0,121 ℃滅菌20 min,用于C.glutamicum轉化子的恢復和生長。
種子培養基(g/L):豆粕水解1.25,(NH4)2SO42,蔗糖 25,KH2PO41, MgSO4·7H2O 0.4,乙酸銨 6,FeSO40.01,MnSO40.01,煙酰胺 0.005,味精 0.1,硫胺素0.000 2,生物素0.000 5,121 ℃滅菌20 min,500 mL三角瓶裝液量50 mL,30 ℃、100 r/min培養16 h。
發酵培養基(g/L):葡萄糖 40,(NH4)2SO436,MgSO4·7H2O 1.5,FeSO40.02,MnSO40.02,KH2PO41,K2HPO41,玉米漿 35,甜菜糖蜜 12,煙酰胺 0.008,甜菜堿0.05,硫胺素0.000 45,生物素0.000 85,CaCO340,pH 7.0,115 ℃滅菌10 min。5 mL種子液轉接到裝有50 mL發酵培養基的500 mL的搖瓶中,30 ℃、 100 r/min發酵40 h。
1.1.4 PCR引物序列
基于NCBI數據庫中C.glutamicumATCC 13032基因組序列設計用于各基因敲除框的擴增和驗證引物,基于StreptococcusmutansJH1005基因組設計用于擴增置換基因的引物。如表1所示。
1.2 實驗方法
1.2.1 胞內NADPH合成相關基因的敲除載體pK18-mobsacB-Δzwf、pK18mobsacB-ΔmalE的構建
根據NCBI中C.glutamicumATCC 13032基因序列設計zwf基因上下游以及中間引物,引物序列如表1。提取C.glutamicumATCC 13032基因組為模板,分別以zwf-L-F/zwf-L-R和zwf-R-F/zwf-R-R為引物PCR,獲得分別在3’端和5’端有相同限制性內切酶酶切位點的PCR產物zwf-L、zwf-R。將膠回收后的基因片段zwf-L和重組自殺型質粒載體pK18mobsacB通過HindIII和SalI雙酶切,純化后將zwf-L和線性化質粒pK18mobsacB22 ℃過夜酶連,轉化E.coliJM109,經抗性平板篩選,挑取轉化子菌落PCR,選取較亮條帶對應的單菌落液體培養后提取質粒酶切驗證,構建質粒pK18mobsacB-Δzwf-L。用同樣的方法將基因片段zwf-R連接至質粒pK18mobsacB-Δzwf-L上構建重組自殺型質粒pK18mobsacB-Δzwf。通過相同的方法構建質粒pK18mobsacB-ΔmalE。
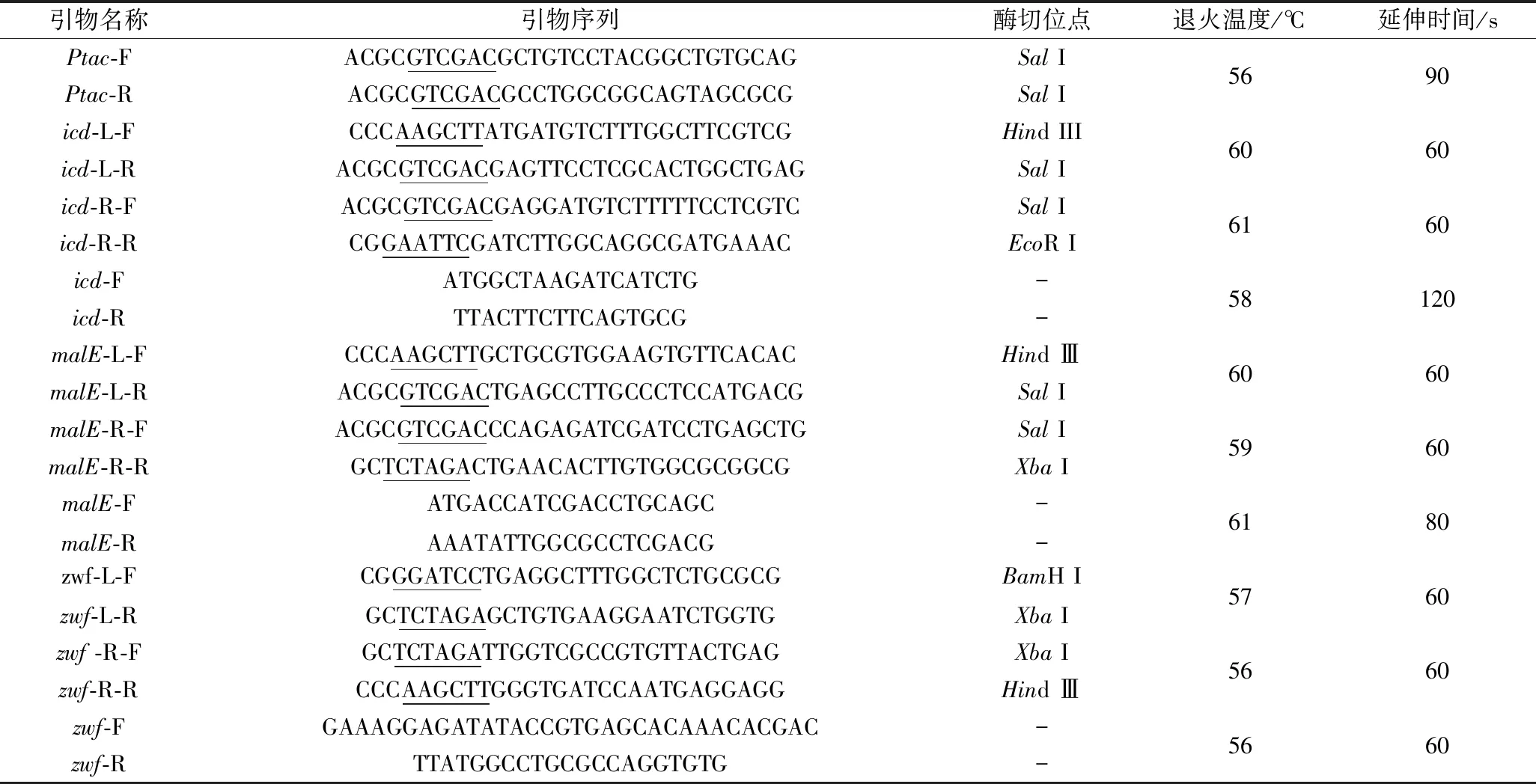
表1 PCR擴增所需引物序列Table 1 Primer sequences required for PCR amplification
注:下劃線為酶切位點。
1.2.2 胞內NADPH合成相關基因的置換載體pK18mobsacB-ΔicdCg::icdSm的構建
通過1.2.1中的方法構建基因敲除質粒pK18mobsacB-ΔicdCg。
根據NCBI中StreptococcusmutansJH1005全基因組核酸序列中的icd基因序列,在其基因上下游分別加入限制性內切酶EcoR I和XhoI酶切位點序列并在上游加入谷氨酸棒桿菌SD識別序列GAAAGGAGATATACC,并將組合好的序列提交給通用生物系統(安徽)有限公司進行合成,獲得含有目的基因的重組質粒pUC57-icdSm。
采用限制性內切酶EcoR I和XhoI酶切重組質粒pUC57-icdSm。隨后采用膠回收試劑盒回收icdSm片段。將icdSm片段與經相同限制性內切酶酶切后的C.glutamicum-E.coli穿梭表達質粒pDXW-8相連構建重組質粒pDXW-8-icdSm。
根據表達質粒pDXW-8基因序列設計包含啟動子Ptac和終止子rrnBT1T2基因上下游引物,引物序列如表1。以pDXW-8-icdSm模板,以Ptac-F/Ptac-R為引物進行PCR,獲得Ptac-icdSm-rrnBT1T2表達框。隨后將純化后的PCR產物Ptac-icdSm-rrnBT1T2與pK18mobsacB-ΔicdCg用SalI單酶切,酶切產物純化后酶連,獲得在icdCg左右同源臂間插入icdSm基因,構建基因置換質粒K18mobsacB-ΔicdCg::icdSm。
1.2.3 重組菌C.glutamicumLYSΔzwfΔmalEΔicdCg::icdSm的構建
1.2.4 胞內輔酶Ⅱ NADP(H)的測定[16]
收集400~500萬細菌,加入0.9 mL酸性(堿性)提取液,超聲破碎1 min(200W,超聲2 s,停1 s),蓋緊后煮沸5 min,冰浴中冷卻后,10 000×g4 ℃離心10 min,取上清液200 μL至另一新的離心管中,加入等體積的堿性(酸性)提取液使之中和,10 000×g、4 ℃離心10 min,取上清。隨后,以試劑盒NADP/NADPH Quantification Colorimeteric Kit特異性檢測NADP+和NADPH,并計算NADPH /NADP+。
1.2.5 胞內輔酶Ⅰ NAD(H)的測定[16]
收集500萬細菌,加入0.5 mL酸性(堿性)提取液,超聲破碎1 min(200W,超聲2 s,停1 s), 蓋緊后煮沸5 min,冰浴中冷卻后,10 000×g4 ℃離心10 min,取上清液200 μL至另一新的離心管中,加入等體積的堿性(酸性)提取液使之中和,10,000×g、4 ℃離心10 min, 取上清。隨后,以試劑盒NAD/NADH Quantification Colorimeteric Kit特異性檢測NAD+和NADH,并計算NADH/NAD+。
1.2.6 重組菌C.glutamicumLYSΔzwfΔmalEΔicdCg::icdSm和出發菌C.glutamicumLYS胞內ATP、ADP、AMP的測定[17]
利用0.6 mol/L HClO4(PCA)抽提重組菌C.glutamicumLYSΔzwfΔmalEΔicdCg::icdSm和出發菌C.glutamicumLYS胞內ATP、ADP和AMP。隨后,采用HPLC技術對抽提液進行分析,測定胞內ATP、ADP和AMP的濃度。將50 mL細胞樣品從培養物中取出,立即在液氮中冷凍60 s,并儲存在-20 ℃。通過HPLC測量細胞內ATP的濃度。為了提取ATP,將10 mL 的0.6 mol/L HClO4加入到細胞沉淀中并混合用磁力攪拌器徹底攪拌10 min。將混合物以10 000×g離心10 min以收集上清液。將另外10 mL的0.6 mol/L HClO4加入到沉淀中,充分混合10 min,離心后收集上清液。將兩部分上清液在25 mL容量瓶中混合并用0.6 mol/L HClO4補足至25 mL。取10 mL所制備的溶液,并用0.8 mol/L KOH將pH值調節至7.0。在4 ℃保持30 min后,通過過濾從孔中除去晶體KClO4(孔徑=0.22 μm),然后在應用HPLC柱之前用磷酸鹽緩沖液(pH 7.0)稀釋至25 mL。 HPLC分析的進樣量為10 μL。使用80%10 mmol/L KH2PO4(pH 7.0)和20%甲醇的混合物作為流動相,流速為1.2 mL/min。紫外檢測器的波長設置為260 nm, 柱溫控制在25 ℃。
1.2.7 分析方法
菌體濃度測定:將發酵液稀釋26倍,測定562 nm處的吸光值。
葡萄糖測定:將發酵液離心(9,000×g,2 min)除去菌體和CaCO3,然后稀釋100倍,經生物傳感分析儀SBA-40C(山東省科學院生物研究所)測定[18]。
氨基酸含量測定:發酵液中氨基酸含量的測定采用氨基酸自動分析儀[19]。
搖瓶發酵出發菌株C.glutamicumLYS和重組菌株C.glutamicumLYS ΔzwfΔmalEΔicdCg::icdSm,發酵期間定時取樣測定發酵液實時葡萄糖含量、菌體量和L-賴氨酸產量并繪制過程曲線。
2 結果與分析
2.1 表達載體和重組菌株的構建
2.1.1 胞內NADPH合成相關基因的敲除載體pK18mobsacB-Δzwf、pK18mobsacB-ΔmalE的構建
E.coliJM109 pK18mobsacB-Δzwf擴大培養后提取質粒分別用BamH I +XbaI和Hind Ⅲ +XbaI雙酶切驗證基因zwf左右同源臂,從酶切電泳結果圖1泳道3、4可以看出,酶切后的左右同源臂片段大小約為900 bp, 與理論大小相符。E.coliJM109 pK18mobsacB-ΔmalE擴大培養后提取質粒分別用Hind Ⅲ +SalI和XbaI +SalI雙酶切驗證基因malE左右同源臂,從酶切電泳結果圖1泳道1、2可以看出,酶切后的左右同源臂片段大小約為900 bp,與理論大小相符。

M-DL5000 Marker;1-質粒pK18mobsacB-ΔmalE Xba I+Sal I雙酶切產物,右同源臂;2-質粒pK18mobsacB-ΔmalE HindIII + Sal I雙酶切產物,左同源臂;3-質粒pK18mobsacB-Δzwf HindIII + Xba I雙酶切產物,右同源臂;4-質粒pK18mobsacB-Δzwf BamH I + Xba I雙酶切產物,左同源臂圖1 質粒pK18mobsacB-Δzwf、質粒pK18mobsacB-ΔmalE左右同源臂雙酶切驗證Fig.1 Enzyme digestion of the recombinant plasmid pK18mobsacB-Δzwf and pK18mobsacB-ΔmalE
2.1.2 胞內NADPH合成相關基因的置換載體pK18mobsacB-ΔicdCg::icdSm的構建
E.coliJM109 pK18mobsacB-ΔicdCg::icdSm擴大培養后提取質粒,由于驗證時沒有合適的酶切位點,只能通過質粒PCR驗證基因icdCg左右同源臂和置換基因icdSm,從PCR產物電泳結果圖2泳道1可以看出基因icdSm片段大小約為1 600 bp,與理論大小相符,從泳道2、3可以看出,左右同源臂片段大小約為900 bp,與理論大小相符。

M-DL5000 Marker;1-質粒pK18mobsacB-ΔicdCg::icdSm icdSm基因PCR產物;2-質粒pK18mobsacB-ΔicdCg::icdSm右同源臂PCR產物; 3-質粒pK18mobsacB-ΔicdCg::icdSm左同源臂PCR產物圖2 質粒pK18mobsacB-ΔicdCg::icdSm各片段PCR驗證Fig.2 PCR analysis of the recombinant plasmid pK18mobsacB-ΔicdCg::icdSm
2.1.3 重組菌C.glutamicumLYSΔzwfΔmalEΔicdCg::icdSm的構建
將篩選到的C.glutamicumLYSΔzwf、C.glutamicumLYSΔzwfΔmalE、C.glutamicumLYSΔzwfΔmalEΔicdCg::icdSm菌株擴大培養后提取基因組后,分別對應以zwf、malE、icd基因上下游引物進行PC鑒定篩選完成二次同源重組菌株,從PCR產物電泳結果圖3泳道1可以看出基因icd置換片段大小約為2 391 bp,與理論大小相符,泳道2、3可以看出基因malE、zwf敲除型殘余片段大小約為599、895 bp,與理論大小相符。各PCR產物測序比對結果也與實驗設計吻合,最終鑒定正確的轉化子命名為C.glutamicumLYSΔzwfΔmalEΔicdCg::icdSm。

M-DL5000 Marker;1-基因icd置換型PCR產物;2-基因malE敲除型PCR產物;3-基因zwf敲除型PCR產物圖3 重組菌PCR驗證Fig.3 PCR analysis of the recombinant strain
2.2 重組菌和出發菌胞內腺嘌呤核苷酸(ATP、ADP和AMP)、吡啶核苷酸(NAD+、NADH、NADP+和NADPH)的變化
為了確定C.glutamicumLYS在敲除zwf、malE基因和替換icd基因后的重組菌是否成功阻斷胞內NADPH的合成量,本文對于出發菌和重組菌搖瓶發酵40 h后對其胞內吡啶核苷酸含量進行測定,具體數據如表2。

表2 重組菌和出發菌胞內吡啶核苷酸(NAD+、NADH、NADP+和NADPH)含量Table 2 Contents of intracellular pyridine nucleotides (NAD+, NADH, NADP+ and NADPH) in recombinant and original bacteria
注:a:單位是μmol/g DCW。表3同。
從表2可以看出重組菌C.glutamicumLYSΔzwfΔmalEΔicdCg::icdSm胞內NADH含量由出發菌的1.97 μmol/g DCW增加到2.73 μmol/g DCW,胞內的NADH/NAD+提高了53.84%,NADPH含量降低了89.51%,在該酶的催化下和出發菌株相比不再合成NADPH而合成更多的NDAH,說明自身NADP+-依賴型異檸檬酸脫氫酶基因(icdCg)替換成變形鏈球菌(Streptococcusmutans)NAD+-依賴型異檸檬酸脫氫酶基因(icdSm),實現了輔酶依賴型的轉變;胞內NADPH含量降低至0.15 μmol/g DCW,明顯低于出發菌胞內NADPH含量1.43 μmol/g DCW。胞內的NADPH/NADP+降低了93.13%,胞內少量的NADPH,可能是由于阻斷了胞內NADPH的合成,NAD(H)/NADP(H)失去平衡后,NAD激酶開始發生作用,使得少量的NADH被NAD激酶催化形成NADPH[24]。谷氨酸棒桿菌體內還有多個涉及NADPH代謝反應沒有發現,如呼吸鏈中NADPH氧化酶的作用[25]。表3可以看出胞內ATP含量降低了13.1%,阻斷了胞內NADPH的合成,即敲除了PPP途徑和改變了異檸檬酸脫氫酶的輔酶依賴型后不再合成NADPH, NADPH的急劇降低致使經過電子傳遞鏈氧化生成的ATP也在一定程度上被削弱,胞內ADP和AMP無法及時轉換成ATP,均有一定程度累積,擾亂了胞內腺嘌呤核苷酸平衡。

表3 重組菌和出發菌胞內腺嘌呤核苷酸 (ATP、ADP和AMP)含量Table 3 Contents of intracellular adenine nucleotides (ATP, ADP and AMP) in recombinant and original bacteria
2.3 阻斷胞內NADPH合成途徑對葡萄糖消耗速率和菌體生長的影響
由圖4可知出發菌和重組菌的生長趨勢基本保持一致,但重組菌的生長較為緩慢,生長速率偏低,在相等OD562相同的接種量下,菌體延滯期基本相同,而菌體的對數生長期延長約4 h。由圖5可知在發酵過程的12~24 h,主要在菌體的對數生長期重組菌的葡萄糖代謝能力明顯弱于出發菌株。
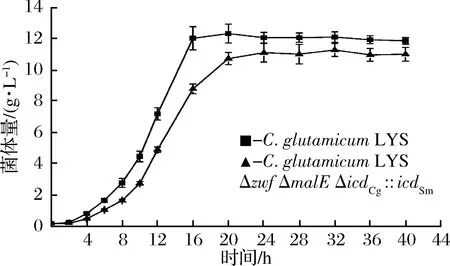
圖4 出發菌和重組菌株菌體生長曲線Fig.4 Growth curve of the oringinal and recombinant strains

圖5 出發菌和重組菌株葡萄糖消耗速率Fig.5 Glucose consumption rate of the oringinal and recombinant strains
由于戊糖磷酸途徑是糖代謝的第二條重要途徑同樣也是細胞產生還原力(NADPH)的主要途徑,敲除了合成NADPH的PPP途徑,胞內NADPH銳減,減緩胞內各種需要NADPH的合成反應,如氨基酸、脂類及核苷酸等細胞組成物質的合成,對細胞正常生長和代謝有重要影響,發酵結束后重組菌菌體量與出發菌相比降低了4.36%。阻斷了葡萄糖進入PPP途徑,葡萄糖代謝途徑減少致使葡萄糖的代謝減緩。胞內的NADH/NAD+提高了53.84%,更高的NADH/NAD+比率不利于糖酵解的進行,致使重組菌的葡萄糖代謝能力進一步削弱。胞內NAD激酶發生作用,形成少量的NADPH優先供應細胞生長,致使阻斷胞內L-賴氨酸合成通路中NADPH的合成,對菌體生長影響相對較小,而對L-賴氨酸產量等代謝產物影響較大。菌體生長也因葡萄糖代謝減緩而受到一定程度的影響。而NADPH也會影響胞內微環境,如NAD(H/+)狀態和ATP含量,由于ATP可調節胞內pHi,提高細胞對酸脅迫的適應力,NADPH可通過NADPH氧化酶阻止胞內羥基等活性氧簇(ROS)的形成,提高細胞對氧脅迫的適應力[26]。而阻斷了NADPH合成途徑導致胞內微環境擾亂,有可能間接性的影響到菌體生長和葡萄糖代謝。
2.4 阻斷胞內NADPH合成途徑對L-賴氨酸合成的影響
由圖6可知,40 h發酵結束后出發菌C.glutamicumLYS發酵液中L-賴氨酸含量為6.5 g/L,重組菌C.glutamicumLYS ΔzwfΔmalEΔicdCg::icdSm,L-賴氨酸產量為0.8 g/L,與出發菌相比,阻斷胞內NADPH合成途徑胞內的NADPH降低了89.51%,L-賴氨酸產量降低了87.69%。

圖6 搖瓶培養條件下出發菌和重組菌株L-賴氨酸產量Fig.6 L-lysine yield of the oringinal and recombinant strains in shake flask culture
敲除了葡萄糖-6-磷酸脫氫酶編碼基因使得PPP途徑失活“C”通量減少,TCA循環“C”通量增加,蘋果酸酶編碼基因敲除和異檸檬酸脫氫酶由于基因置換導致輔酶依賴型的改變使得胞內NADPH含量降低至0.15 μmol/g DCW。在發酵12 h后,出發菌和重組菌的菌體量開始出現較大的差距,同時L-賴氨酸含量也開始出現很明顯的差距,結果表明此時由于重組菌無法滿足菌體生長和L-賴氨酸合成所需的NADPH,導致菌體生長緩慢,菌體量減少,并且在L-賴氨酸合成途徑中谷草轉氨酶、天冬氨酸半醛脫氫酶、二氫吡啶二羧酸還原酶、內消旋二氨基庚二酸脫氫酶或琥珀酰-氨基-吡咯酮轉氨酶需要以NADPH為輔酶,合成1分子L-賴氨酸需要4分子的NADPH,NADPH的嚴重缺乏,加劇了L-賴氨酸的產量的降低。大量研究表明,L-賴氨酸產量的增加與增加PPP途徑“C”通量和減少TCA循環“C”通量密切相關。
2.5 阻斷胞內NADPH合成途徑對副產物合成的影響
很多氨基酸的合成都依賴于NADPH,為了進一步研究阻斷胞內NADPH合成途徑對發酵副產物如有機酸和其他氨基酸的影響,發酵結束后測定發酵液中有機酸和副產物氨基酸含量。
由表4可以看出由于阻斷了胞內NADPH合成途徑出發菌C.glutamicumLYS和重組菌C.glutamicumLYS ΔzwfΔmalEΔicdCg::icdSm發酵液中的副產物氨基酸明顯降低,纈氨酸、亮氨酸、異亮氨酸、蘇氨酸、谷氨酸較出發菌株分別降低了76.94%、79.22%、 83.09%、80.87%、62.25%,纈氨酸、亮氨酸、異亮氨酸、蘇氨酸、谷氨酸的合成都需要提供輔因子NADPH,致使合成產量都明顯降低,而由于合成谷氨酸所需要的輔因子NADPH相對較低,對其生物合成的影響相比其他氨基酸而言相對較低,而賴氨酸合成需要更多的輔因子NADPH,致使其產量降低的最為明顯。

表4 重組菌和出發菌發酵液中副產物氨基酸和有機酸含量Table 4 The concentration of amino acid and organic acid in fermentation of recombinant and original bacteria
而重組菌較低的菌體量對發酵過程中各種氨基酸副產物的積累也有一定程度的削弱。發酵液中的有機酸測定結果表明,發酵液中的丙酮酸和乳酸含量與出發菌相比分別增高了41.55%、15.73%,是由于阻斷了胞內NADPH合成途徑后,極大削弱了菌株內以NADPH為輔酶的氨基酸合成,如以丙酮酸為前體,合成的丙氨酸族氨基酸,和經過丙酮酸流向三羧酸循環后以草酰乙酸為前體合成的天冬氨酸族氨基酸,使得丙酮酸等代謝途徑上游的中間產物得以累積。
3 結論
該研究中對L-賴氨酸產生菌C.glutamicumLYS進行基因工程改造,構建了重組自殺型質粒pK18mobsacB-Δzwf、pK18mobsacB-ΔmalE用于敲除葡萄糖-6-磷酸脫氫酶編碼基因zwf和蘋果酸酶編碼基因malE,克隆來源于Streptococcusmutans的NADP+-依賴型異檸檬酸脫氫酶(編碼基因icdCg)通過重組自殺型質粒pK18mobsacB-ΔicdCg::icdSm替換谷氨酸棒桿菌NAD+-依賴型異檸檬酸脫氫酶(編碼基因icdSm),依次將重組質粒電轉化至C.glutamicumLYS,篩選并獲得完成二次同源重組的菌株C.glutamicumLYSΔzwfΔmalEΔicdCg::icdSm,該重組菌阻斷了谷氨酸棒桿菌中NADPH合成的相關途徑。由于異檸檬酸脫氫酶是三羧酸循環中將檸檬酸催化生成異檸檬酸,通過基因置換以改變其輔因子依賴型,本研究中用來源于Streptococcusmutans的異檸檬酸脫氫酶替換谷氨酸棒桿菌原有的異檸檬酸脫氫酶。對出發菌和重組菌進行搖瓶發酵實驗,對兩種不同的輔因子NADPH水平下胞內氧化還原輔因子(NAD+、NADH、NADP+和NADPH)和能量輔因子(ATP、ADP和AMP)進行分析和比較,NADPH含量較出發菌降低了89.51%,NADPH/NADP+降低了93.13%,NADH含量提高了38.58%,NADH/NAD+提高了53.84%,ATP含量降低了13.1%。重組菌的葡萄糖代謝能力明顯弱于出發菌株,菌體的生長也相對緩慢,進入穩定期的時間延長,最終菌體量與出發菌相比降低了4.36%。L-賴氨酸產量降低了87.69%, 纈氨酸、亮氨酸、異亮氨酸、蘇氨酸、谷氨酸合成量也都明顯降低,而在胞內積累了大量副產物,如丙酮酸、乳酸等。當阻斷胞內NADPH的合成途徑時,胞內微環境如氧化還原水平等受到擾動,此時菌體生長受到影響,而對L-賴氨酸產量的改變尤為明顯。為進一步解析輔因子NADPH調控L-賴氨酸產生菌胞內微環境的生理機制奠定基礎。
