4-羥氨基-α-吡喃酮甲酰胺類似物的3D-QSAR及分子對接研究
2019-07-08 09:35:52仝建波秦尚尚
原子與分子物理學報 2019年3期
仝建波, 秦尚尚, 雷 珊, 王 洋
(1.陜西科技大學化學與化工學院, 西安 710021; 2.教育部輕化工助劑化學與技術重點實驗室, 西安 710021)
1 引 言
丙型肝炎病毒(Hepatitis C virus,HCV)是一種單股正鏈的RNA病毒,也是慢性肝病的主要原因之一[1]. 根據2017年中國疾病預防控制中心報道,全世界約有1.3~2.1億人感染丙型肝炎病毒(HCV),85%的患者會發展成為慢性肝炎,最終將導致肝硬化、肝功能衰竭或肝癌[2],嚴重影響患者的身體健康和生活質量,但至今還未開發出有效的丙型肝炎疫苗和治療措施[3,4]. HCV感染性疾病的治療目標是患者獲得持續病毒學應答(SVR,如SVR24為治療結束至隨訪24周時PCR檢測血清HCV RNA為陰性). 國際上公認的標準治療方案(SOC)是聚乙二醇-干擾素α(PEG-IFNα)聯合利巴韋林(RBV)[5]. 但是實踐證明該治療方案不僅具有嚴重的不良反應,治療周期偏長,而且大部分患者不能耐受,接受48周完整治療患者并沒有達到很高的SVR獲得率(基因1和4型HCV感染患者SVR獲得率僅為40%~50%)[6]. 因此,研究新的治療方案迫在眉睫.
直接抗病毒藥物(DAA)是指特異性作用于維持HCV生命周期所必需的酶或蛋白質的藥物. DAA的研發與應用為HCV感染的治療帶來新希望. DAA主要包括NS3/4A蛋白酶抑制劑、NS5B聚合酶抑制劑及NS5A抑制劑[7,8]. 應用DAA并且聯合其他技術應用于臨床試驗發現,這樣做不僅可以擺脫干擾素的輔助治療,而且用藥也比較方便,不良反應大大降低. 因此,尋找更合適的抗病毒結構是有意義的,它可以為成功解決SOC缺陷、成為有前景的抗HCV藥物奠定一定的理論基礎.
三維定量構效關系[9-11]是一種應用廣泛的設計藥物分子的方法,它可以有效地把分子結構與其理化性質、結構參數、生物活性等結合起來. 三維定量構效關系中最具代表性的方法有CoMFA(Coparative molecular field analysis)與CoMSIA(Coparative molecular similarity index analysis),即比較分子場方法和比較分子相似性方法[12,13]. 本論文采用第二代CoMFA方法Topomer CoMFA[14,15]對42個4-羥氨基-α-吡喃酮甲酰胺類似物進行3D-QSAR研究,得到該類化合物結構與其生物活性之間的3D-QSAR模型,用于新型HCV抑制劑的設計. 相比于傳統方法,本文所采用的Topomer CoMFA得到的模型,具有更好的穩定性和預測能力,還省去了分子疊合的步驟,具有花費時間短,重復性高等優勢. 因此利用該方法可以快速建立模型進行預測、分析和評價,為小分子抑制劑的結構優化提供理論基礎.
2 方法與步驟
2.1 數據集的選擇
42個4-羥氨基-α-吡喃酮甲酰胺類似物的分子結構和活性值如表1所示,均取自文獻報道[16]. 把化合物的pEC50(-logEC50)值作為活性數據進行模型建立,它是由半數抑制濃度EC50轉化過來的. 把具有試驗數據的42個化合物大致按照4:1的比例隨機分為訓練集和測試集,其中訓練集包括33個化合物,測試集包括9個化合物. 用訓練集來進行模型的建立,用測試集進行模型的驗證.
2.2 Topomer CoMFA模型建立
Topomer CoMFA是CoMFA與Topomer的聯合技術,能夠在短時間內準確、快速構建3D-QSAR模型. 采用Topomer技術將整個配體分子切割成兩個或兩個以上的小片段,所切割形成的小片段會自動生成三維構象的碎片,碎片根據一定的經驗規則進行調整,生成Topomer模型. 本文中所用到化合物的結構是由Sketch Molecule繪制出來的,它是Tripos公司Sybyl2.0-X軟件包中的一個模塊. 將繪制出的化合物結構進行優化,優化過程采用Powell能量梯度法、Tripos立場等,其中能量收斂設定為0.005 kcal/mol,優化次數設定為1000次,分子荷載電荷為Gasteiger-Huckel電荷,其余參數均為默認值.
對訓練集中33個化合物的結構進行切割,將活性最高的21號化合物作為模板分子,采用如圖1所示的切割方式進行切割,分別生成了R1和R2基團,隨后軟件會自動識別并且切割其他的分子結構,最后對R1和R2基團周圍的立體場和靜電場自動進行計算,并把它作為自變量,以EC50的負對數(pEC50)為建模響應值,采用偏最小二乘(Partial least squares method,PLS)回歸分析法[17]進行建模來表示化合物活性與分子場之間的關系,生成3D-QSAR模型. 采用留一法交互驗證[18,19]評價模型的內部預測能力,然后利用所建立的Topomer CoMFA模型預測9個測試集化合物的活性,以此評價模型的外部預測能力.
2.3 虛擬篩選
本文中的虛擬篩選采用Sybyl2.0-X軟件中的Topomer Search技術,它采用構象的獨立性及的Topomer相似性進行完整分子、側鏈或骨架的篩選. Topomer Search[20]結果中主要包含兩項:Topomer距離和R基團的活性貢獻值,其可從數據庫中篩選并測出相應R基團貢獻值. 通常在分子相似程度允許范圍內,貢獻值越大的基團越有考慮價值.
表1 4-羥氨基-α-吡喃酮甲酰胺類似物的分子結構及其活性值
Table 1 The structure and activity values of the 4-hydroxyamino α-pyranone carboxamide analogues

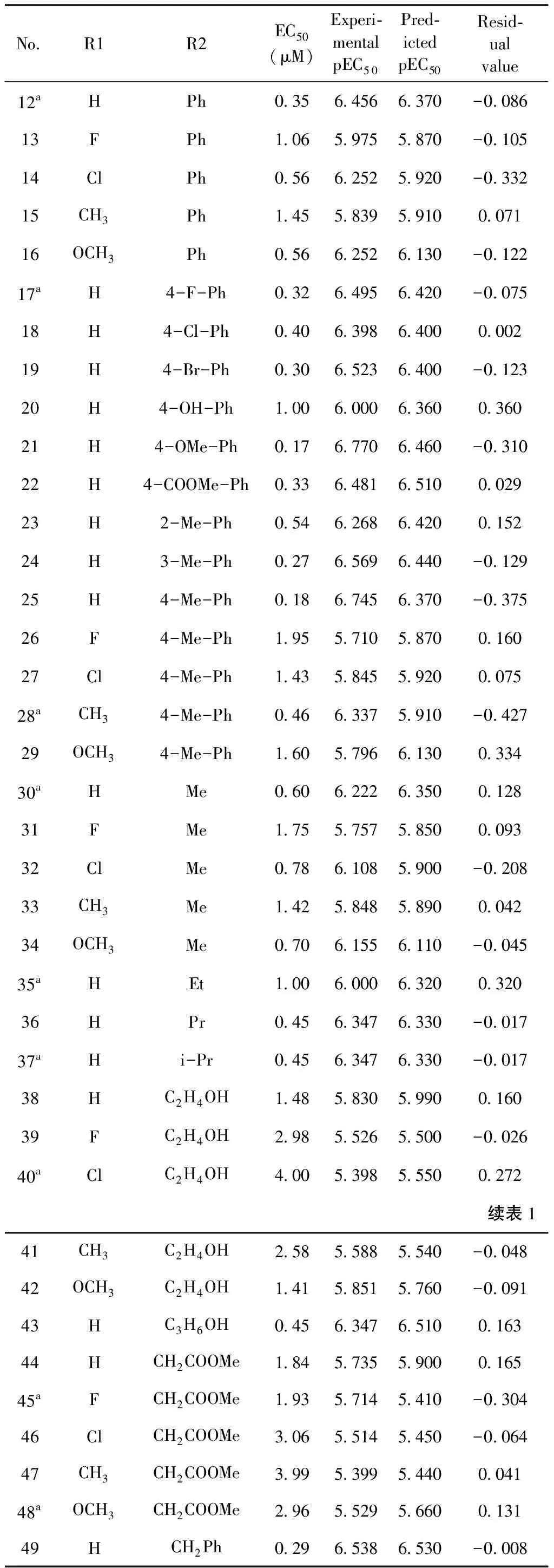
No.R1R2EC50(μM)Experi-mentalpEC50Pred-ictedpEC50Resid-ualvalue12aHPh0.356.4566.370-0.08613FPh1.065.9755.870-0.10514ClPh0.566.2525.920-0.33215CH3Ph1.455.8395.9100.07116OCH3Ph0.566.2526.130-0.12217aH4-F-Ph0.326.4956.420-0.07518H4-Cl-Ph0.406.3986.4000.00219H4-Br-Ph0.306.5236.400-0.12320H4-OH-Ph1.006.0006.3600.36021H4-OMe-Ph0.176.7706.460-0.31022H4-COOMe-Ph0.336.4816.5100.02923H2-Me-Ph0.546.2686.4200.15224H3-Me-Ph0.276.5696.440-0.12925H4-Me-Ph0.186.7456.370-0.37526F4-Me-Ph1.955.7105.8700.16027Cl4-Me-Ph1.435.8455.9200.07528aCH34-Me-Ph0.466.3375.910-0.42729OCH34-Me-Ph1.605.7966.1300.33430aHMe0.606.2226.3500.12831FMe1.755.7575.8500.09332ClMe0.786.1085.900-0.20833CH3Me1.425.8485.8900.04234OCH3Me0.706.1556.110-0.04535aHEt1.006.0006.3200.32036HPr0.456.3476.330-0.01737aHi-Pr0.456.3476.330-0.01738HC2H4OH1.485.8305.9900.16039FC2H4OH2.985.5265.500-0.02640aClC2H4OH4.005.3985.5500.272續表141CH3C2H4OH2.585.5885.540-0.04842OCH3C2H4OH1.415.8515.760-0.09143HC3H6OH0.456.3476.5100.16344HCH2COOMe1.845.7355.9000.16545aFCH2COOMe1.935.7145.410-0.30446ClCH2COOMe3.065.5145.450-0.06447CH3CH2COOMe3.995.3995.4400.04148aOCH3CH2COOMe2.965.5295.6600.13149HCH2Ph0.296.5386.530-0.008

50FCH2Ph1.185.9286.0400.11251ClCH2Ph0.686.1676.090-0.07752CH3CH2Ph0.916.0416.0700.02953OCH3CH2Ph0.576.2446.2900.046
Note:aTest set (測試集化合物)
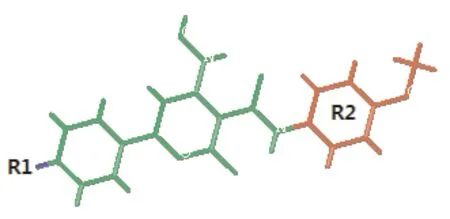
圖1 模板分子的切割方式Fig. 1 Cutting mode for template molecules
本文所選取的數據庫為ZINC(2012)數據庫中的Drug-like類,共有130000個分子,通過篩選得到具有高活性貢獻值的R1和R2基團. 其中,Topomer CoMFA距離值設置為185,其他參數以默認值為準.
2.4 分子對接
本文采用Sybyl2.0-X軟件中的Surflex-dock模塊來完成對接,對接所使用的蛋白酶晶體結構來源于PDB數據庫,晶體結構的PDB ID為:1ADG,對接模式為Surflex-dock(SFXC),在進行對接之前需對晶體結構進行預處理,具體包含以下步驟:加氫、加電荷、抽取原有配體、除去水分子和其他殘基及末端殘基的處理. 在準備小分子的過程中,將配體的輸出構象設置為20個,其他參數基本默認,同時選擇原配體結構作為參比分子,進行小分子配體與大分子蛋白的對接與打分,打分結果用Total-Score、Crash和Polar表示[21]. 通常情況下,Total-Score為總的打分函數,表示受體與配體的親和能力,打分越高越好;Crash絕對值越接近零,表示配體與受體對接時的不適當程度越小. Polar為極性函數得分,當結合位點位于分子表面時,打分越高越好;當結合位點位于分子內部時,打分越低越好.
3 結果與討論
3.1 Topomer CoMFA建模結果與評價
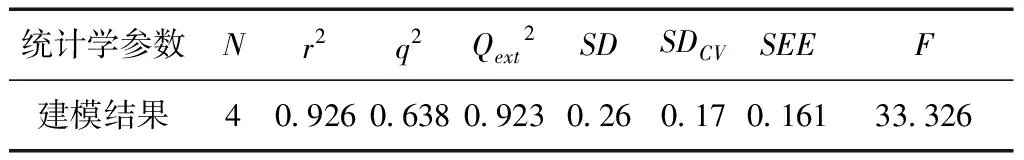
表2 Topomer CoMFA建模結果
注:N為主成分數;r2為擬合復相關系數;q2為交互驗證復相關系數;Qext2為外部驗證復相關系數;SD為擬合標準偏差;SDCV為交互驗證標準偏差;SEE為標準估計偏差;F為Fisher驗證值.

圖2 42個化合物活性的實驗值與預測值的線性回歸圖Fig. 2 The linear regression of the actual value and predictive value of 42 compounds
3.2 Topomer CoMFA等勢圖分析
圖3所示即為21號模板分子的Topomer CoMFA三維等勢圖,其中圖3a和圖3b分別表示R1基團的立體場三維等勢圖和靜電場三維等勢圖,圖3c和圖3d分別表示R2基團的立體場三維等勢圖和靜電場三維等勢圖. 在圖3a和圖3c中,綠色的區域表示引入體積較大的取代基有利于化合物活性的提高,黃色的區域表示引入體積較小的取代基有利于化合物活性的提高;在圖3b和圖3d中,紅色區域表示引入帶負電性取代基有利于化合物活性的提高,藍色區域表示引入帶正電性取代基有利于化合物活性的提高.
在圖3a中,R1取代基附近有大片的黃色區域以及一小塊綠色區域,表明在此處引入體積較小的取代基有利于化合物活性的提高. 例如化合物25(pIC50=6.745)在R1取代基的位置上用H取代了化合物26、27、28和29上(pEC50=5.710,5.845,6.337,5.796)的F,Cl,-CH3及-OCH3取代基,活性明顯提高. 在圖3c中,在苯環的附近被大塊的綠色區域所包圍,這表明在此處適宜引入體積較大的基團,有助于提高化合物的生物活性. 例如,化合物12(pEC50=6.456)的R2位置上為-Ph,它的活性明顯大于R2位置上為-Me,-Et,-Pr的化合物30,35和36(pEC50=6.222,6.000,6.347).
在圖3b中,R1取代基附件有大片的藍色區域,表明此處不宜引入電負性大的化合物. 例如,化合物25(pEC50=6.745)的生物活性明顯大于化合物26(pEC50=5.710),就是由于化合物25的R2取代基位置為電負性較小的-H,而化合物26的R2取代基位置為電負性較強的-F. 在圖3d中,苯環的附近有大片的藍色區域,表明此位置基團電負性減小,其活性反而增大. 例如,-OH的電負性大于-CH3,故化合物38(pEC50=5.830)的活性小于化合物36(pEC50=6.000)的活性. 少量的紅色色區域出現在苯環的C-4位置,表明此處引入電負性大的基團有利于活性的提高. 例如,-Br的電負性明顯大于-H,故19號化合物(pEC50=6.523)的活性明顯高于12號化合物(pEC50=6.456).
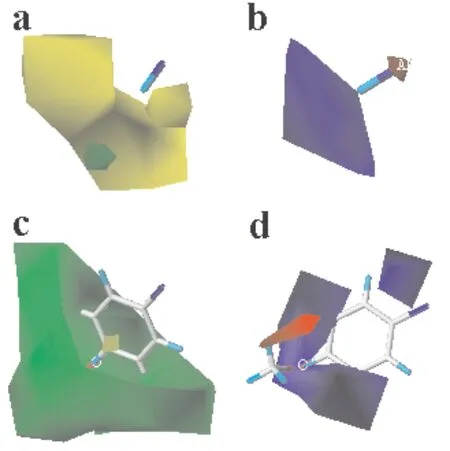
a: R1立體等勢圖 (Steric field map of R1); b:R1靜電等勢圖(Electrostatic field map of R1); c: R2立體等勢圖(Steric field map of R2); d: R2靜電等勢圖(Electrostatic field map of R1).圖3 Topomer CoMFA三維等勢圖Fig. 3 The results of Topomer CoMFA
3.3 基于Topomer Search的分子設計
利用Topomer Search技術挑選出來的2個R1基團和16個R2基團對21號模板分子中的相應基團進行替換,設計得到了32個新的化合物. 在Sybyl2.0-X軟件包中的Sketch Molecule模塊繪制出這32個新分子的結構,然后對其進行優化,優化方法與之前的42個樣本分子相同. 最后,將優化好的新化合物通過之前所建立的Topomer CoMFA模型進行活性預測,以下表3所示即為32個新設計的化合物結構及其活性預測值.
從表3中可得知,有30個新設計的化合物的預測活性值比模板分子高,說明新設計的化合物理論上具有一定的病毒抑制作用,可以作為抗艾滋病新藥的候選化合物. 從新設計分子的結構中可以觀察到,在R1取代基的位置引入體積較小、電負性較大的基團,且在R2取代基的位置引入體積較大、電負性較大的基團,所以化合物的生物活性有所提高. 除了這些原因,這2個化合物活性值低還可能是由于:篩選出來的R1與R2基團雖然單獨都具有較高的貢獻值,但是合在一起組成化合物時卻不相匹配;還有可能是因為重組形成的新化合物內部有不利的相互作用阻礙其活性增大. 以上解釋均符合Topomer CoMFA模型等勢圖分析.
3.4 分子對接結果分析
在進行分子對接前,需要對分子對接方法準確性和可靠性進行驗證. 通過Surflex-dock將晶體結構中的配體分子抽取出來作為參照分子,然后再重新與晶體結構1ADG進行對接,所得到的結果如下圖所示. 圖4a所示即為共晶配體與1ADG的對接模式圖,從圖中可以看出對接口袋為一條狹長的通道,位于晶體的中間位置. 圖4b為共晶配體與1ADG的氫鍵作用圖,我們可以從圖中觀察到共晶配體與氨基酸殘基ASP223,ILE368,LYS228,GLY202,ARG47,VAL268等形成氫鍵作用,以上結論即可說明該方法能夠有效的進行分子對接研究.
表3 新設計化合物結構與預測活性值
Table 3 Structures and predictive activity values of new design compounds
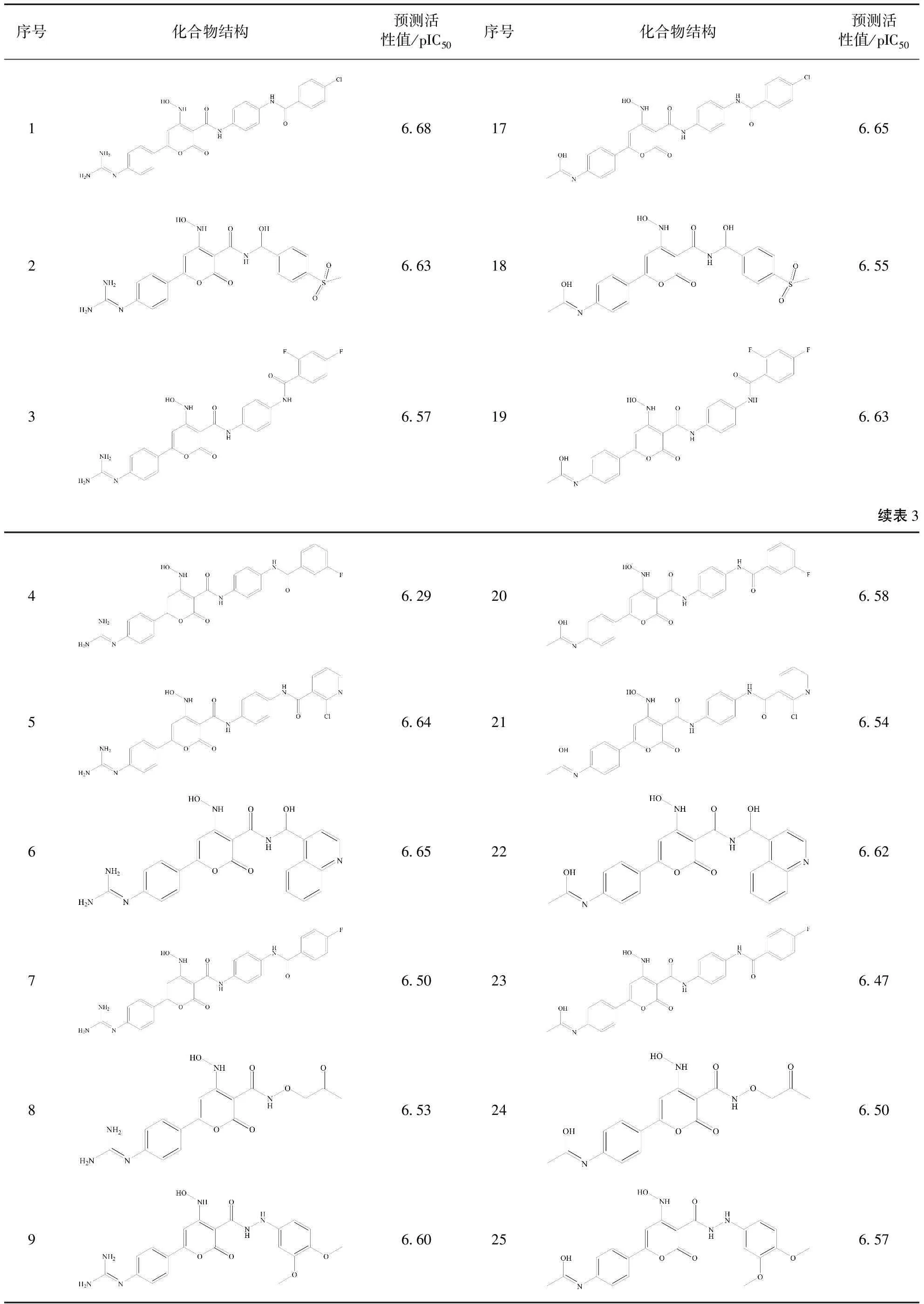
序號化合物結構預測活性值/pIC50序號化合物結構預測活性值/pIC5016.68176.6526.63186.5536.57196.63續表346.29206.5856.64216.5466.65226.6276.50236.4786.53246.5096.60256.57
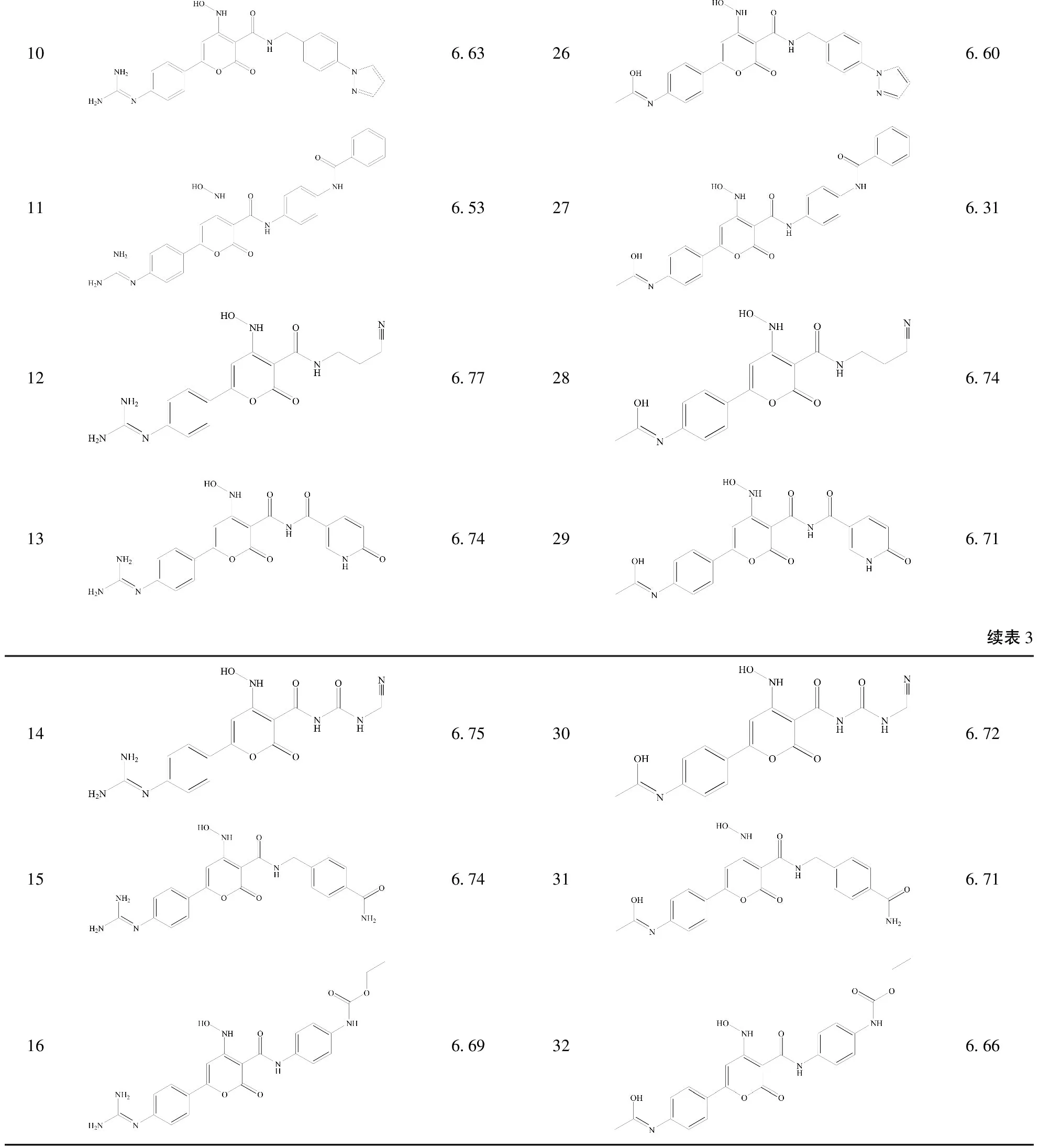
106.63266.60116.53276.31126.77286.74136.74296.71續表3146.75306.72156.74316.71166.69326.66

圖4 (a) 共晶配體與1ADG對接模式(綠色部分為共晶配體,紫色線狀為氨基酸殘基);(b) 共晶配體與1ADG的氫鍵作用(中間棒狀表示共晶配體,紅色虛線表示氫鍵,綠色棒狀表示氨基酸殘基)Fig. 4 (a) The docking mode between reference ligand and 1ADG; (6) The hydrogen bond interaction between reference ligand and 1ADG.
在本文中,訓練集中的化合物和新設計的分子都被用來進行分子對接研究. 我們選取訓練集化合物中活性最高的21號模板分子進行對接,氫鍵作用結果如圖5a所示,配體六元環上sp2雜化的O原子與A鏈氨基酸殘基ARG47中的H原子形成氫鍵,距離為2.052 ?;配體中sp3雜化的H原子與A鏈氨基酸殘基ILE368中的O原子形成氫鍵,距離為1.833 ?;配體中sp3雜化的O原子與A鏈氨基酸殘基GLY201中的H原子形成氫鍵,距離為1.997 ?. 總打分函數(Total-Score)、配體對接進受體時的不適當程度(Crash)和極性打分函數(Polar)分別為5.2934、-1.0786和3.1900.
為了更進一步驗證新設計的化合物的抑制效果,將新設計的32個分子分別對接到其蛋白受體上,以研究其結合機制,方法同訓練集分子. 所有新設計分子的總打分函數(Total-Score)如表4所示. 由表可知有17個分子總打分函數高于模板分子,我們選取分數最高的10號,31號和32號分子進行研究. 如圖5b、c、d分別表示新設計的化合物10、31、32與晶體結構1ADG的氫鍵相互作用圖. 在圖5b中,配體10號分子中sp3雜化的O原子與A鏈氨基酸殘基LYS228中的H原子形成氫鍵,其距離為1.826 ?;配體中sp3雜化的H原子與A鏈氨基酸殘基ASP223中的O原子形成2個氫鍵,距離分別為2.168 ?和2.095 ?. 總打分函數(Total-Score)、配體對接進受體時的不適當程度(Crash)和極性打分函數(Polar)分別為7.7828、-0.9319和3.2247. 圖5c中31號配體中sp2雜化的N原子與A鏈氨基酸殘基GLY202中的H原子形成2個氫鍵,距離分別為2.569 ?和2.093 ?;配體中sp3雜化的H原子與A鏈氨基酸殘基ILE368中的O原子形成氫鍵,其距離為2.301 ?;配體中sp3雜化的H原子與A鏈氨基酸殘基ASP223中的O原子形成2個氫鍵,距離分別為2.224 ?和2.290 ?;配體中sp3雜化的O原子與A鏈氨基酸殘基LYS228中的H原子形成氫鍵,其距離為2.434 ?. 總打分函數(Total-Score)、配體對接進受體時的不適當程度(Crash)和極性打分函數(Polar)分別為7.1290、-2.3703和4.1662. 在圖5d中,32號配體與受體共形成7個氫鍵作用,分別為:配體中sp3雜化的H原子與A鏈氨基酸殘基ASP223中的O原子形成2個氫鍵,距離分別為2.038 ?和2.558 ?;配體中sp2雜化的N原子與A鏈氨基酸殘基GLY202中的H原子形成2個氫鍵,距離分別為2.573 ?和2.169 ?;配體中sp3雜化的H原子與A鏈氨基酸殘基ILE368中的O原子形成氫鍵,其距離為2.168 ?;配體中sp3雜化的O原子與A鏈氨基酸殘基ARG369中的H原子形成2個氫鍵,距離分別為2.442 ?和2.752 ?. 總打分函數(Total-Score)、配體對接進受體時的不適當程度(Crash)和極性打分函數(Polar)分別為6.8815、-2.8634和4.5604. 這些氫鍵作用使配體小分子與大分子蛋白結合更加穩定,所以對接結果具有一定的可靠性. 但是有一些新設計的化合物例如3號、7號、14號、17號等均具有較高的活性值,但是總打分函數卻不高,這些化合物也是無意義的. 雖然使用Topomer Search可以得到具有高貢獻值的某些片段,但將片段整合后形成的化合物卻不具備良好的結果,這可能是因為碎片和支架在空間位置或能量上不匹配,證明了分子的局部優化具有有界性,片段和支架之間的合理匹配是藥物設計的重要條件.

表4 新設計分子的總打分函數

圖5 氫鍵相互作用圖(a、b、c、d分別表示21號模板分子、新設計分子10號、31號、32號與1ADG的氫鍵作用圖)Fig. 5 a The hydrogen-bond interaction between template molecular 21 and 1ADG. b the hydrogen-bond interaction between new design compound 10 and 1ADG. c the hydrogen-bond interaction between new design compounds 31 and 1ADG. d the hydrogen-bond interaction between new design compounds 32 and 1ADG (the red dotted lines represent the hydrogen bonding)
4 結 論
本文采用Topomer CoMFA方法對42個4-羥氨基-α-吡喃酮甲酰胺類似物進行了三維定量構效關系的研究,得到了穩定性和預測能力都比較好的3D-QSAR模型. 采用Topomer Search技術在ZINC數據庫對R基團進行虛擬篩選,將篩選出來的基團重新組合,設計了32個新的化合物,進一步預測其活性,有30個分子活性高于模板分子. 利用模板分子與新設計的分子進行對接,探究配體和受體蛋白之間的作用關系,對接結果表明小分子與大分子蛋白的氨基酸殘基ARG47、ILE368和GLY201可以形成氫鍵,說明所建立的模型值得信任,可為設計高活性抗HCV分子提供理論參考.
