高溫成膠可降解聚合物凝膠堵漏劑的研制與評價
2019-07-10 08:45:56郭永賓顏幫川黃熠李磊蔣官澄鄧正強
鉆井液與完井液 2019年3期
關鍵詞:體系
郭永賓,顏幫川,黃熠,李磊,蔣官澄,鄧正強
(1.中海石油(中國)有限公司湛江分公司,廣東湛江524057;2.中國石油大學(北京),北京 102200)
近年來,有機凝膠堵漏劑、無機凝膠堵漏劑和橋接堵漏劑等堵漏劑雖然取得了一定進展,但堵漏成功率低,對提高地層承壓能力有限,堵漏成本高,難以滿足惡性漏失地層的封堵要求[1-3]。聚合物凝膠堵漏工藝是在地面將單體和交聯劑混合,混合后具有較低的黏度,通過鉆桿泵入漏失地層,依靠漏失地層溫度,在一定時間里形成具有一定強度的凝膠,從而封堵漏層。普通的凝膠堵漏存在高溫成膠時間短、凝膠強度低及成膠后難以破膠等問題[1-8]。高溫交聯可降解聚合物凝膠:①采用樹脂包覆引發劑控制高溫成膠時間。②通過天然大分子接枝多乙烯基單體交聯丙烯酰胺,具有高溫下成膠時間可控、成膠強度高和成膠后可破膠,不存在堵漏劑與漏失通道尺寸級配的問題,有效地封堵不同孔隙和縫洞。
1 交聯凝膠堵漏劑的研制與表征
1.1 可降解交聯劑
1.1.1 分子結構設計
在自由基聚合中,有機交聯劑N,N-二甲基雙丙烯酰胺,通常具有多點引發的特點,然而有機交聯劑鍵能大,穩定性好,一旦交聯成密集三維網架結構,高溫下難以破膠[9-11]。因此,在其分子結構設計時主要考慮以下幾點:①必須含有多點引發官能團,具有交聯位置;②交聯劑本身在高溫下具有可降解性,即含有可高溫降解的分子結構;③水溶性好,可充分溶解在清水中;④與其它成分的配伍性能好。
1.1.2 合成步驟
通過季銨化反應,利用甲基丙烯酸酐將明膠改性,得到可聚合交聯劑[12-14]。在三口燒瓶中加入10 g明膠和100 mL蒸餾水中,水浴加熱至60 ℃,保溫1 h至天然大分子全部溶解,以1 mL/min速率滴加6 mL甲基丙烯酸酐,滴加后反應3 h。反應過程中保持氮氣環境。反應結束后,使用透析膜透析7 d,后冷凍干燥得到可降解交聯劑DGCL。其合成原理見圖1。
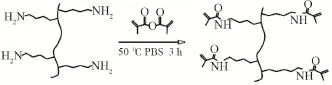
圖1 明膠改性交聯劑的合成原理
1.1.3 核磁共振表征
將提純后的明膠和甲基丙烯酸酐改性明膠分別溶于重水中,進行1HNMR核磁共振分析(見圖2)。由圖2可知,明膠改性后在化學位移5.4 ppm和5.6 ppm處出現特征峰,為甲基丙烯酸酐官能團中丙烯質子特征峰;在化學位移1.87 ppm處的峰,為甲基丙烯酸酐官能團中甲基質子特征峰。說明甲基丙烯酸酐成功接枝到明膠上。
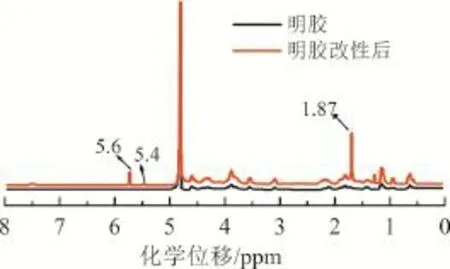
圖2 明膠改性前后1H-NMR分析實驗
1.2 延遲引發劑
1.2.1 設計思路
熱熔膠是一種具有熱塑性的智能聚合物,在高溫下成流動態,當溫度降低至玻璃態溫度以下時恢復成不可流動態。利用該特點,在高溫下將流動態熱溶膠與引發劑混合,冷卻至室溫后,得到熱熔膠引發劑膠囊,從而實現高溫延遲釋放的效果。
1.2.2 合成
在三口燒瓶中加入20 g熱熔膠,油浴加熱至150 ℃,保溫30 min至熱熔膠充分熔化成流動態液體,后加入2 g引發劑,攪拌均勻后倒入冷水中冷卻成固體,烘干、粉碎得到延遲引發劑CIT-1。
1.3 凝膠堵漏劑基本配方
基于自由基聚合,以丙烯酰胺聚合鏈為骨架鏈,以改性明膠為交聯劑。熱熔膠引發劑在高溫下熔化釋放自由基引發聚合,得到可降解凝膠體系,其基本原理見圖3。A523為陰離子聚丙烯酰胺,分子量為100萬。凝膠體系基本配方如下。
清水+2% A523+7% AM+0.1% CIT-1+0.04 %DGCL+5% NaCl
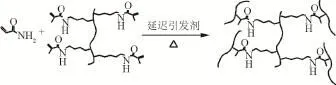
圖3 凝膠體系的成膠原理
2 抗高溫可降解凝膠堵漏劑性能評價
2.1 成膠時間的確定
2.1.1 CIT-1濃度對成膠時間的影響
引發劑濃度對自由基聚合速率起著決定性的作用[15]。在高溫條件下,研究了不同濃度CIT-1對聚丙烯酰胺凝膠體系成膠時間的影響,結果見圖4。由圖4可知,隨著CIT-1濃度的增大,體系成膠時間逐漸下降,當CIT-1濃度小于0.2%時,成膠時間大于6 h,當CIT-1濃度達到1%時,成膠時間在1 h左右。分析其可能的原因是CIT-1濃度過低時,引發劑釋放的自由基過少,凝膠體系中的單體未得到完全引發,導致成膠時間過長,隨著CIT-1濃度的增加,自由基濃度增大,凝膠體系單體得到引發,聚合成具有足夠鏈長的三維網架結構,從而快速形成凝膠。
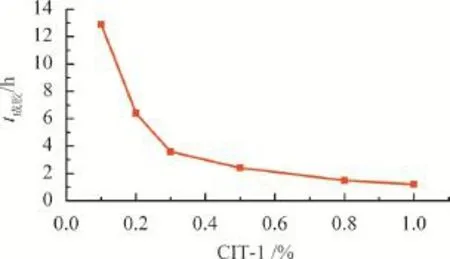
圖4 CIT-1濃度對聚丙烯酰胺凝膠體系成膠時間的影響
2.1.2 溫度對成膠時間的影響
不同溫度下引發劑的半衰期隨著溫度的上升,半衰期呈指數下降[16-17]。在保證配方一致的條件下,分別研究了80~150 ℃下聚丙烯酰胺凝膠體系的成膠時間(見圖5)。由圖5可知,隨著溫度的增加,成膠時間逐漸下降。其可能的原因是:①隨著溫度的增加熱熔膠膠囊熔化速率增加,導致引發劑釋放速率加快,從而縮短成膠時間;②隨著溫度的增加,引發劑半衰期下降,進一步縮短成膠時間。

表1 溫度對聚丙烯酰胺凝膠體系成膠時間的影響
2.2 力學性能測試
凝膠體系中交聯劑濃度的高低決定了凝膠交聯密度,從而影響凝膠的力學性能。參照標準GB/T 528—2009/ISO 37∶2005,使用啞鈴狀試樣裁刀制作成長75 mm、寬4 mm、厚2 mm的啞鈴狀凝膠片。斷裂延伸率計算公式為式(1)。

式中:δ為斷裂延伸率;L0為試樣原始標距,mm;L為試樣斷裂時標線間的距離,mm。
拉伸強度計算公式為式(2)。

式中,σ為試樣拉伸強度,MPa;F為最大拉伸力,N;a為試樣寬度,mm;h為試樣厚度,mm。
選取明膠改性交聯劑,通過電子拉力試驗機做拉伸實驗(見表2)。由表2可知,交聯劑DGCL加入量為0.04%時伸長最好,斷裂延伸率達1279%,拉伸強度為0.0425 MPa,交聯劑DGCL-1加入量為0.03%時,拉伸強度最大為0.0436 MPa,斷裂伸長率為798%。由于凝膠堵漏劑對凝膠強度和拉伸要求高,結合斷裂延伸率和拉伸強度,交聯劑DGCL加量為0.04%最佳。
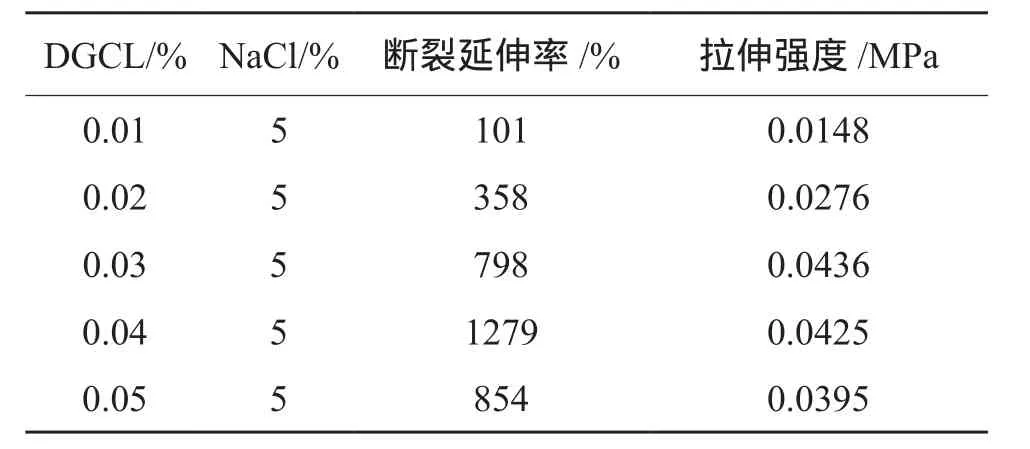
表2 不同DGCL加量下聚丙烯酰胺凝膠體系的拉伸實驗
2.3 砂床堵漏實驗
使用71型高溫高壓失水儀進行砂床堵漏實驗。將4~8目100 g石英砂加入高溫高壓失水儀中,搗實。分別將配制好的堵漏漿PHPA-Cr3+、PHPAPEI和P(AM-DGCL)倒入儀器中(PHPA為部分水解聚丙烯酰胺,Cr3+為金屬鉻離子交聯劑,PEI為聚乙烯亞胺),150 ℃保溫4 h后進行承壓堵漏實驗,結果見圖5。由圖5可知,P(AM-DGCL)凝膠體系相比金屬離子Cr3+、交聯凝膠PHPA-Cr3+和PHPA-PEI具有更好的承壓堵漏能力。P(AMDGCL)凝膠可承壓6.9 MPa以上,且累計漏失量小于30 mL。PHPA-Cr3+和PHPA-PEI交聯凝膠承壓分別為2.76和4.83 MPa,且漏失量是P(AMDGCL)凝膠的10倍以上。其主要的原因是P(AMDGCL)凝膠是有機交聯,其交聯機理是交聯劑本身具有可聚合多官能團,在自由基聚合過程中參與聚合,交聯密度大,凝膠強度高,而金屬離子Cr3+和PEI交聯劑是將已有的聚合物PHPA進行官能團交聯,一方面交聯鍵能更低,另一方面由于已有聚合物的纏繞形成更大的空間位阻導致交聯密度低,凝膠強度弱,從而堵漏效果差。
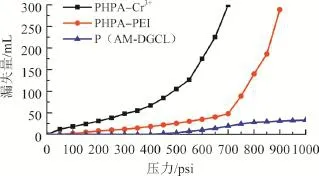
圖5 不同交聯凝膠體系承壓堵漏實驗結果
2.4 破膠性實驗
儲層漏失堵漏后,為保證儲層的生產能力,需要將堵漏漿破膠返排[18-21]。對此,進行了凝膠破膠實驗(見圖6)。稱取一定量已成膠的凝膠堵漏劑,加入2%過硫酸銨,攪勻倒入老化罐,在滾子爐中滾動加熱一定時間后,取出凝膠,過篩稱量剩余凝膠。計算破膠率。破膠率計算公式為式(3)。

式中:D為破膠率率;m0為破膠后剩余凝膠質量,g;m為破膠前凝膠質量,g。
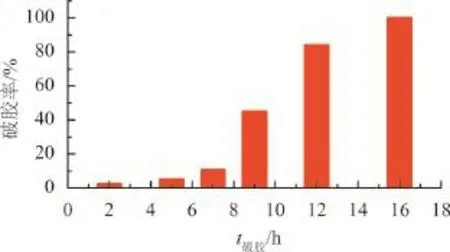
圖6 堵漏凝膠破膠率隨破膠時間的變化情況
由圖6可知,該凝膠體系在150 ℃下,破膠時間9 h后破膠率達40%以上,當破膠時間為16 h,破膠率達95%以上,基本破膠完全,滿足儲層反排要求。其破膠的主要機理是明膠在高溫和氧化的條件下發生一定程度的降解,導致凝膠交聯密度大幅度下降,從而實現破膠。
3 結論
1.合成了甲基丙烯酸酐改性明膠交聯劑,通過參與自由基聚合形成交聯密度大的高強度凝膠。
2.基于熱熔膠的熱塑性原理,合成了可控緩釋引發劑膠囊,實現了成膠時間的延長,滿足現場施工要求。
3.合成的凝膠體系具有良好的可破膠性,加入2%過硫酸銨破膠劑,150 ℃破膠16 h,破膠率達95%以上,保證了儲層堵漏后良好的反排能力。
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
