TiO2改性提高LiNi0.8Co0.1Mn0.1O2正極材料的電化學及儲存性能
2019-07-20 01:49:10林桂仙張春曉韋偉峰
中國材料進展 2019年5期
關鍵詞:改性
林桂仙,韓 博,黃 群,張春曉,韋偉峰
(中南大學 粉末冶金國家重點實驗室,湖南 長沙 410083)
1 前 言
長期以來,鋰離子二次電池作為高效便捷的電化學儲能系統,在手機、數碼相機、筆記本電腦、智能手表等領域獲得了廣泛應用[1,2]。近些年來由于市場和政府的雙重鼓勵和支持,鋰離子二次電池也在電動汽車(EVs)、智能電網等市場展現出了巨大的潛力[3-5]。高鎳系層狀三元氧化物正極材料LiNi1-xMxO2(x<0.4,M=Mn,Co,Al等)因具有高比容量(170~200 mAh·g-1)、低成本、環境友好等優點,被認為是下一代動力電源、大型儲能設備的最佳選擇之一[6-8]。但該材料仍存在循環穩定性差、儲存條件苛刻、熱穩定性差、脹氣現象嚴重等問題,并隨著Ni含量的升高而變得更加嚴重[9-11]。首先,由于Ni2+的半徑(0.69 ?)與Li+的半徑(0.74 ?)相近,高鎳系層狀三元氧化物正極材料在合成過程中極易產生Ni2+/Li+混排,從而形成尖晶石/巖鹽結構,阻礙Li+的脫/嵌,造成材料容量損失[12,13]。其次,與低鎳系正極材料相比,高鎳系層狀三元氧化物正極材料在深度脫Li+狀態下Ni4+含量更高,而Ni4+具有強氧化性,容易與電解液發生副反應釋放氣體,引發安全問題,同時導致材料表面形成非活性的NiO相,使得界面阻抗增加[13-18]。除此之外,高鎳系層狀三元氧化物正極材料在存儲過程中易與空氣中的CO2和H2O反應生成LiOH、Li2CO3等殘鋰附著于材料的表面,同時伴隨著活性鋰的損失與NiO相的形成,導致材料層狀結構受到破壞;而且在充放電循環過程中,殘鋰的存在也會阻礙離子和電子的遷移并加劇與電解液之間的副反應[19-22]。
針對上述缺陷,研究人員做了大量的改性研究,其中表面包覆和體相摻雜被認為是兩種行之有效的策略[2,15,23]。一方面,通過在活性材料表面均勻地涂覆Al2O3[20,24,25]、TiO2[26-28]、ZrO2[2,29,30]、SiO2[16,31]等保護層,能夠將其與外界環境隔離,不但能減少與CO2/H2O之間的副反應,而且可以有效抑制電解液中產生的HF對活性材料的侵蝕。另一方面,許多研究證明摻雜Na+ [32,33]、Ti4+ [34,35]、Al3+ [21,36]、Zr4+ [37,38]、F-[39]等離子能夠穩定材料的結構、降低陽離子混排、提高Li+擴散系數,從而改善材料的電化學性能。然而,關于表面包覆與體相摻雜協同作用對LiNi0.8Co0.1Mn0.1O2電化學行為和儲存性能的研究還很有限。
因此本實驗利用鈦酸四丁酯水解與800 ℃高溫復燒相結合的方法,同時實現了對LiNi0.8Co0.1Mn0.1O2的TiO2表面包覆與Ti4+體相摻雜改性,提高了其循環穩定性和儲存性能。并通過一系列測試,討論了TiO2表面包覆與Ti4+體相摻雜對LiNi0.8Co0.1Mn0.1O2材料結構和電化學性能的影響。
2 實 驗
2.1 材料制備
原始的LiNi0.8Co0.1Mn0.1O2樣品合成步驟如下:將NiSO4、CoSO4、MnSO4按照摩爾比nNi2+∶nCo2+∶nMn2+=8∶1∶1,配制成總濃度為2 mol/L的混合鹽溶液,再配制2 mol/L的Na2CO3溶液與0.24 mol/L的氨水混合的堿溶液。將混合鹽溶液、堿溶液以合適的速率并流滴加至反應釜中,快速攪拌,同時精確控制反應釜的溫度及pH值穩定在7~8。反應一段時間后,將反應所得的沉淀物洗滌、過濾后在120 ℃真空條件下干燥12 h即可得到(Ni0.8Co0.1Mn0.1)CO3前軀體。接著將所得的碳酸鹽前軀體置于馬弗爐中在空氣氣氛下450 ℃預處理3 h,得到(Ni0.8Co0.1Mn0.1)O2前軀體。最后,稱取一定量的LiOH·H2O(過量3%)與氧化物前驅體充分研磨混合均勻,在氧氣氣氛下810 ℃燒結20 h,最終得到所需原始的LiNi0.8Co0.1Mn0.1O2樣品,命名為811-bare。
采用濕化學法及后續熱處理對LiNi0.8Co0.1Mn0.1O2進行TiO2修飾步驟如下:首先,將32.5 μL的鈦酸四丁酯分散于30 mL的乙醇中,然后將1 g LiNi0.8Co0.1Mn0.1O2正極材料粉末超聲分散在上述溶液中,再逐滴加入1 mL去離子水,以促使鈦酸四丁酯緩慢水解,隨后在60 ℃條件下蒸干溶液,將所得混合物在氧氣氣氛下800 ℃后處理5 h,隨爐冷卻后得到TiO2改性的LiNi0.8Co0.1Mn0.1O2正極材料,命名為811@TiO2。
2.2 電池的組裝與電化學性能測試
將正極材料與導電炭黑、聚偏氟乙烯(PVDF)按照8∶1∶1(質量比)的比例充分混合后溶于N-甲基吡咯烷酮(NMP)中,高速攪拌2 h后將均勻的漿料涂覆于鋁箔上。將涂覆后的鋁箔放置于80 ℃鼓風干燥箱內干燥6~8 h。隨后用手動沖片機將干燥好的極片裁剪成直徑為12 mm的圓片,接著用輥壓機壓實極片后放到120 ℃真空干燥箱內干燥12 h,最終得到可以裝備電池的極片,活性物質面密度大約為1.1 mg·cm-2。最后,在氬氣氣氛的手套箱內組裝CR2016型扣式電池。該電池采用金屬鋰片作為負極,Celgard2400有機物薄膜為隔膜,1 mol/L LiPF6溶于EC/DMC(體積比=1∶1)的混合溶液為電解液。本實驗使用Land電池測試系統進行電化學性能測試。測試采用倍率充放電,充放電電壓范圍為 3.0~4.3 V,測試溫度為28 ℃。長效循環測試參數設置:首先以0.1C(20 mA·g-1)的電流密度充放電1圈,活化電極材料,從第2圈開始,采用2C(400 mA·g-1)的電流密度進行100圈充放電測試,以評估正極材料的結構穩定性。
2.3 材料分析與表征
本文采用場發射掃描電子顯微鏡(Nova Nano SEM230)、透射電子顯微鏡(Tecnai G2 F20)、雙束電子顯微鏡FIB-SEM(Helios Nanolab G3 UC)表征材料的形貌,并結合能譜分析儀(EDX)對各元素的分布進行分析。采用透射電子顯微鏡(Tecnai G2 F20)、X射線衍射儀(XRD)對材料的物相組成和晶體結構進行表征。X射線衍射儀測試條件:以CuKα射線為輻射源,步長0.02 s,停留時間1.2 s,掃描角度范圍10°~80°。并利用GSAS-EXPGUI軟件對XRD結果進行精修,分析材料經改性后晶胞參數的變化。采用原子發射光譜(ICP)來確定材料組成成分。同時利用X射線光電子能譜(XPS)對材料表面的離子價態和含量進行表征。測試采用結合能為284.8 eV的C 1s作為標準進行校正。
3 結果與討論
圖1a是LiNi0.8Co0.1Mn0.1O2原始樣和TiO2改性LiNi0.8-Co0.1Mn0.1O2樣的XRD圖譜。從圖中可以看出,兩者都具有純相的α-NaFeO2層狀結構,屬于R-3m空間群。同時,(108)/(110)和(006)/(102)這兩組峰分裂明顯,說明合成的正極材料均具有良好的結晶性[23]。XRD結果表明微量TiO2改性并沒有改變材料的主體結構,但由于TiO2含量過低,其所對應的衍射峰強度較弱,無法被檢測到。從圖1b和1c中(003)與(104)衍射峰的局部放大圖可以看出,與原始樣相比,TiO2改性后樣品的(003)與(104)衍射峰均向低角度偏移,由布拉格方程2dsinθ=nλ(d為晶面間距,θ為入射角,n為反應級數,λ為波長)可知,改性后的LiNi0.8Co0.1Mn0.1O2的晶面間距變大。原始樣和TiO2改性樣的組分由ICP-AES確定,分別為Li1.023Ni0.797-Co0.104Mn0.099O2和 Li1.016Ni0.795Co0.103Mn0.102Ti0.0098O2,基本符合實驗預期。
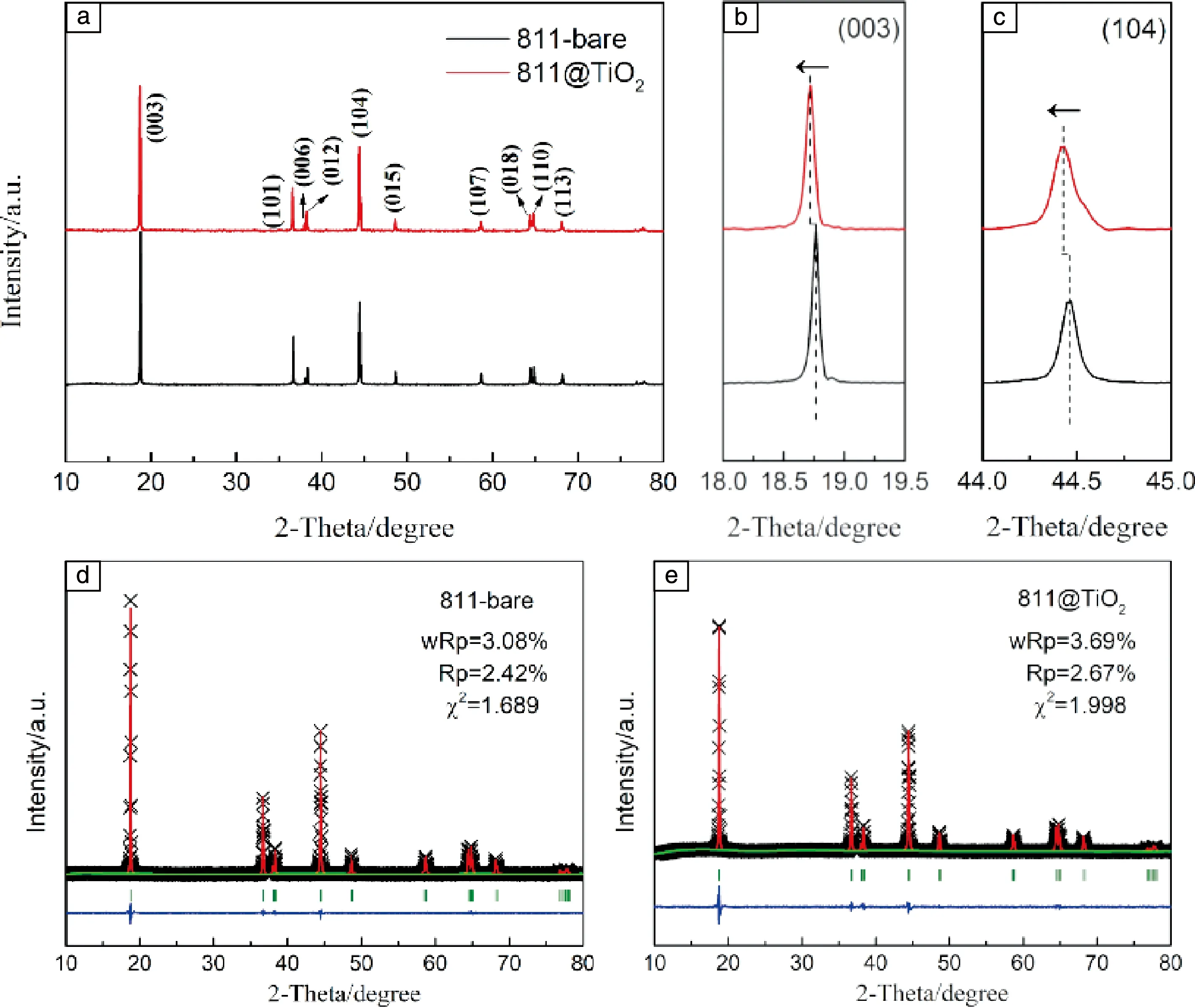
圖1 811-bare原始樣和TiO2改性811-bare樣品的XRD圖譜(a),(003)和(104)峰局部放大圖(b,c),811-bare原始樣和TiO2改性811-bare樣品的Reitveld精修結果圖譜(d,e)Fig.1 X-ray diffraction patterns of the 811-bare and 811@TiO2 cathode materials (a),enlarged regions for the (003)and (104)peaks of the XRD patterns(b,c),reitveld refinement of the XRD patterns for the 811-bare and TiO2 modified samples(d,e)
為了進一步定量分析晶胞參數變化,對兩個樣品的XRD圖譜進行了Rietveld精修理,其結果如圖1d和1e以及表1所示。與未改性樣相比,TiO2改性樣的晶胞參數略微增大,這主要是由于少量的Ti4+(0.605 ?)進入LiNi0.8Co0.1Mn0.1O2的晶格中,并取代了半徑較小的過渡金屬[34,40-42]。與此同時,原始樣與TiO2改性樣品的I(003)/I(104)的比值分別為1.8484和1.7953,表明TiO2改性會造成陽離子混排加劇。Liu等[41]通過對比TiO2包覆與Ti4+摻雜對LiNi0.8Co0.2O2材料結構與性能的影響發現,TiO2包覆并不會影響材料的晶體結構,而Ti4+摻雜則會使晶胞參數擴張,也會加劇鋰鎳混排。因此可以推測,Ti4+成功地進入晶格體內,并為了維持電荷平衡,使部分Ni3+還原為Ni2+,進而加劇鋰鎳混排[13]。為了驗證TiO2改性樣表面TiO2包覆層的存在及Ti4+的摻雜,采用EDS mapping表征材料截面的元素分布。如圖3c,Ni,Co,Mn這3種元素在截面上均勻分布,而Ti元素則在表面富集,并有一部分在高溫熱驅動的作用下進入材料晶格。

表1 精修后的811-bare原始樣和TiO2改性樣晶胞參數Table 1 Refined crystallographic parameters for the 811-bare and 811@TiO2 samples
圖2a和2b為TiO2改性前后LiNi0.8Co0.1Mn0.1O2正極材料的SEM照片。可以發現,未包覆樣品的一次顆粒表面光滑,棱角分明;而經TiO2改性后,一次顆粒表面變得粗糙,在其表面附著密集的點狀納米顆粒。為了進一步確定TiO2包覆層的形成,采用TEM對原始樣和TiO2改性樣的微觀結構進行表征分析,如圖3a和3b所示。從圖中可以看出,包覆前后的樣品的晶體結構都具有清晰的層狀條紋,晶面間距為0.47 nm,對應六邊形層狀結構(R-3m)的(003)面。與原始樣光潔平整的表面相比,TiO2改性樣表面有一層均勻的TiO2層,其晶面間距為0.23 nm,與銳鈦礦TiO2的(004)晶面一致。可以推測,這層TiO2保護膜將有效地阻止電解液中的HF對LiNi0.8-Co0.1Mn0.1O2正極材料的侵蝕,減少有害副反應的發生,進而改善其循環穩定性[26,27]。

圖2 811-bare原始樣的SEM照片(a),TiO2改性811-bare樣品的SEM照片(b)Fig.2 SEM images of the 811-bare material (a),SEM images of the 811@TiO2 material (b)

圖3 811-bare原始樣的HRTEM照片和對應的FFT照片(a),TiO2改性811-bare樣品的HRTEM照片和紅色區域放大照片(b),TiO2改性811-bare樣的截面EDS元素分布圖(c)Fig.3 HRTEM image and corresponding FFT pattern (inset)of the 811-bare sample (a),HRTEM image of the 811@TiO2 sample and magnified HRTEM image taken from red region (inset)(b),cross-section EDS mappings of an 811@TiO2 secondary particle (c)

圖4c和4d為原始樣和TiO2改性樣以400 mA·g-1電流密度充放電循環時第5,25,45,65,100圈的電壓-放電比容量曲線圖。在循環過程中,原始樣的放電電壓和放電比容量衰減都非常嚴重,而經過TiO2改性的樣品其電壓和放電比容量的衰減都得到了有效的抑制。為了進一步探究TiO2改性后材料循環穩定性提高的原因,對原始樣和TiO2改性樣分別在1,20,50圈循環后進行循環伏安(CV)測試,結果如圖4e和4f所示。兩個樣品都出現了3對氧化還原峰,分別位于3.72,3.90和4.15 V附近,對應原始層狀結構(H1)→單斜相(M)、單斜相(M)→層狀結構(H2)和層狀結構(H2)→層狀結構(H3)這3個相變過程[7,11]。在充放電循環過程中,原始樣的3對氧化還原峰均發生大角度位移,例如,位于3.72 V的氧化峰經過50圈循環后向高電位移動至3.83 V并伴隨著顯著的峰強降低現象,這意味著原始樣在循環過程中,極化嚴重且放電容量衰減劇烈。而對于經TiO2改性的LiNi0.8Co0.1Mn0.1O2樣來說,極化現象明顯小于原始樣,表明Li+脫/嵌的可逆性增強。
對循環1,20,50次后的原始樣和TiO2改性的LiNi0.8Co0.1Mn0.1O2電極進行了EIS測試,結果如圖5所示。兩個樣品的EIS譜圖都由兩個半圓和一條斜線組成,其中在最高頻區位于Zre軸上的截距表示歐姆電阻(Rs),位于高頻區的半圓和低頻區的半圓分別對應SEI膜阻抗(Rsei)和電荷轉移電阻(Rct),而斜線則代表Warburg擴散電阻[44,45]。按照圖5中的等效電路進行擬合,得到的擬合數據結果如表2所示。結合表2和圖5可知,兩個樣品的阻抗值隨著循環的進行而不斷增加,但相對于原始樣,TiO2改性樣阻值的增加更為緩慢。以電荷轉移電阻值Rct為例,經過50圈循環后,原始樣的Rct值由91.24增大至263.7 Ω,而TiO2改性的的樣品的Rct則增加較小(60.91增大至119.8 Ω)。以上現象表明TiO2改性有效地抑制了活性材料與電解液之間的副反應,使LiNi0.8Co0.1Mn0.1O2正極材料的界面更加穩定,更有利于多次循環后鋰離子的脫/嵌和電荷的轉移[26]。

表2 811-bare原始樣和TiO2改性樣的EIS阻抗擬合參數Table 2 Fitting parameters of the Nyquist impedance for the 811-bare and 811@TiO2 samples
高鎳層狀正極材料的儲存性能對其商業化應用至關重要。811-bare原始樣與TiO2改性樣品在空氣中暴露30 d后的電化學性能如圖6a和6b所示。從測試結果看,原始樣LiNi0.8Co0.1Mn0.1O2樣品在30 d暴露后初始放電容量大幅度降低,僅有115.8 mAh·g-1,相當于新鮮樣品的初始放電容量的67.40%。在400 mA·g-1電流密度下100次循環后容量保持率也降低至68.02%。此外,在圖6a中也可以觀察到暴露后原始樣明顯的電壓極化。相比較之下,TiO2改性樣表現出更強的空氣穩定性及循環穩定性。在相同的儲存和測試條件下,30 d暴露后的初始放電容量仍有140.9 mAh·g-1,相當于新鮮樣品的初始放電容量的86.12%,在400 mA·g-1電流密度下100圈循環后容量保持率高達85.31%。圖6c對比了原始樣與改性樣在空氣暴露前后的FTIR圖譜。863和1425 cm-1處的峰位是CO32-的特征峰[46],此處的CO32-主要來源于活性材料與空氣中的CO2反應生成的Li2CO3。顯然,在暴露30 d后,原始樣的CO32-峰比TiO2改性樣更明顯,表明在空氣中暴露后,原始樣的表面退化程度更大。為了進一步探究暴露前后樣品表面的價態變化情況,作者研究組對30 d暴露前后的原始樣和TiO2改性樣進行XPS測試,圖6d顯示暴露前的TiO2改性樣品在458.2和463.9 eV處有明顯的峰位,為Ti4+的特征峰[34],表明改性樣品表面存在TiO2。
暴露前后所有樣品的Ni 2p XPS結果經XPS PEAK軟件擬合,結果如圖6e和6f所示。經過30 d的空氣暴露,原始樣的Ni 2p3/2峰位從855.76顯著降低至855.42 eV,相比較之下,TiO2改性的樣品的Ni 2p3/2峰位只從855.66略微降低至855.54 eV。通過半定量的擬合可以得出,TiO2改性樣經暴露后Ni2+含量(原子百分數)為30.62%,低于原始樣的34.10%。這些現象表明了TiO2改性后的樣品在空氣中暴露后形成較少的NiO巖鹽相[12]。由此可見,TiO2改性可以有效地抑制高鎳正極材料與空氣的副反應,提高高鎳材料的空氣穩定性。

圖6 811-bare原始樣和TiO2改性樣在空氣中暴露30 d后的首圈充放電曲線(a)、循環曲線(b)和FTIR圖譜(c);811-bare原始樣的Ti 2p XPS圖譜(d),811-bare原始樣暴露前后的Ni 2p XPS圖譜(e),TiO2改性樣暴露前后的Ni 2p XPS圖譜(f)Fig.6 Initial charge-discharge profiles (a),cycling performance (b)and FTIR spectra (c)of the 811-bare and the 811@TiO2 materials after being exposed for 30 days in air;XPS spectrums of Ti 2p of the 811@TiO2 materials (d),Ni 2p of the 811-bare materials before and after being exposed (e),Ni 2p of the 811@TiO2 materials before and after being exposed (f)
4 結 論
采用濕化學法及高溫后處理成功地將TiO2均勻包覆在LiNi0.8Co0.1Mn0.1O2表面,并實現了部分Ti4+的體相摻雜,使其循環穩定性及儲存性能均優于原始樣。經過改性,以400 mA·g-1的電流密度循環100圈后LiNi0.8Co0.1Mn0.1O2的容量保持率由80.38%提高到90.77%。在空氣中暴露30 d后的電化學測試中,改性后的LiNi0.8Co0.1Mn0.1O2樣品展現出優異的空氣穩定性,仍具有140.9 mAh·g-1的初始放電比容量,容量保持率為85.31%;而原始樣的初始放電比容量僅為115.8 mAh·g-1,容量保持率下降至68.02%。上述性能的改善一方面歸因于TiO2包覆層,不但抑制了充電狀態下LiNi0.8Co0.1Mn0.1O2正極材料與電解液的副反應,而且減少了正極材料與空氣的直接接觸,進而減少儲存過程中的表面結構退化;另一方面,摻雜入晶格的Ti4+有助于提高材料的結構穩定性,顯著降低了循環過程中的阻抗增加及不可逆相變過程。
猜你喜歡
紡織科學研究(2020年1期)2020-05-21 00:31:06
中國塑料(2016年12期)2016-06-15 20:30:07
中國塑料(2016年2期)2016-06-15 20:30:00
中國塑料(2016年2期)2016-06-15 20:29:59
中國塑料(2016年5期)2016-04-16 05:25:36
廣西林業科學(2016年3期)2016-03-16 05:43:30
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
中國塑料(2015年4期)2015-10-14 01:09:19
