超高效液相色譜一串聯(lián)質(zhì)譜法檢測(cè)公英青藍(lán)合劑中非法添加物金剛乙胺
2019-08-03 07:16:06魏秀麗張志民趙有軒張傳津李有志
山東農(nóng)業(yè)科學(xué) 2019年3期
魏秀麗 張志民 趙有軒 張傳津 李有志
摘要:建立了公英青藍(lán)合劑中非法添加金剛乙胺檢測(cè)的UPLC - MS/MS方法。樣品經(jīng)甲醇提取稀釋后,采用ACQUITY UPLC BEH C18色譜柱為分離柱,質(zhì)譜正離子掃描分析測(cè)定。結(jié)果顯示,金剛乙胺在0.5-50ng/mL范圍內(nèi)線性關(guān)系良好(R2=0.9985);樣品檢出限為2μg/mL,定量限為5μg/mL;在5、25、50、100μg/mL添加水平的回收率為90%~110%,批內(nèi)批間變異系數(shù)均小于10%。本方法靈敏、快速、重現(xiàn)性好,適用于公英青藍(lán)合劑中非法添加金剛乙胺的檢測(cè)。
關(guān)鍵詞:金剛乙胺;公英青藍(lán)合劑;超高效液相一串聯(lián)質(zhì)譜法
Determination of Rimantadine Illegally Added inGongying Qinglan Mixture by UPLC-MS/MS Wei Xiuli, Zhang Zhimin, Zhao Youxuan, Zhang Chuanjin, Li Youzhi
Abstract
A method was established for determination of rimantadine added in Gongying Qinglan Mixtureby UPLC - MS/MS. The sample was extracted and diluted with methanol, the ACQUITY UPLCR BEH C18was used as the separation column, and the determination was conducted by scanning positive ion by massspectrometry. The results showed that the linear relationship of rimantadine was better in the range of 0.5 to50 ng/mL ( R2 =0.9985) . The detection limit was 2 μg/mL, and the quantitation limit was 5μg/mL. The a-nalysis of spiked 5, 25, 50, 100 μg/mL rimantadine showed that the recoveries ranged from 90% t0 110%and the relative standard deviations were below 10%. The experiment showed that the method was reliable,sensitive and reproducible and suitable for the determination of rimantadine illegally added in Gongying Qing-lan Mixture.
Keywords
Rimantadine; Gongying Qinglan Mixture; UPLC - MS/MS
公英青藍(lán)合劑[1]是由蒲公英、大青葉、板藍(lán)根、金銀花、黃芩、黃渤、甘草、藿香、石膏九味藥物組成的滅菌合劑,養(yǎng)禽業(yè)使用頻率較高,具有清熱解毒功效,用于禽類傳染性法氏囊病等病毒病的輔助治療。金剛乙胺在人醫(yī)臨床是一種抗病毒藥物[2],早些年養(yǎng)禽業(yè)臨床獸醫(yī)也使用它防治禽病毒病,但其耐藥性日增且獸藥殘留對(duì)畜產(chǎn)品消費(fèi)者存在較大風(fēng)險(xiǎn)。早在2005年農(nóng)業(yè)部560號(hào)公告已經(jīng)禁止金剛乙胺用于畜禽養(yǎng)殖業(yè),12月又發(fā)布《關(guān)于清查金剛烷胺等抗病毒藥物的緊急通知》,美國(guó)食品藥品監(jiān)督管理局(FDA)也明令禁止在畜禽養(yǎng)殖業(yè)使用此類藥物,禁止金剛烷胺、金剛乙胺抗病毒獸藥的生產(chǎn)和使用,緣由是其作為抗病毒藥物應(yīng)用于獸醫(yī)臨床,缺乏科學(xué)規(guī)范、安全有效的試驗(yàn)數(shù)據(jù)。但少數(shù)不法獸藥企業(yè)在利益的驅(qū)動(dòng)下違規(guī)添加金剛乙胺,嚴(yán)重影響畜禽產(chǎn)品質(zhì)量安全,而獸藥典沒(méi)有相應(yīng)的檢測(cè)方法。本實(shí)驗(yàn)室參考獸藥殘留的檢測(cè)方法[3-6],致力于獸藥中多種非法添加物的檢測(cè)[7,8],研究建立公英青藍(lán)合劑中非法添加物金剛乙胺的超高效液相串聯(lián)質(zhì)譜法檢測(cè)方法,以期為非法添加金剛乙胺的定性、定量測(cè)定提供可靠手段。
1 材料與方法
1.1 儀器
分析天平:感量0.00001g,瑞士Mettler公司;Waters Quattro premier超高效液相一串聯(lián)質(zhì)譜儀、0.22μm濾膜均為Waters公司;離心機(jī),日立。
1.2藥品及試劑
金剛乙胺,批號(hào)100969 - 201101,含量100%,中國(guó)食品藥品檢定研究院。甲酸、甲醇均為色譜純;超純水。
1.3 色譜條件
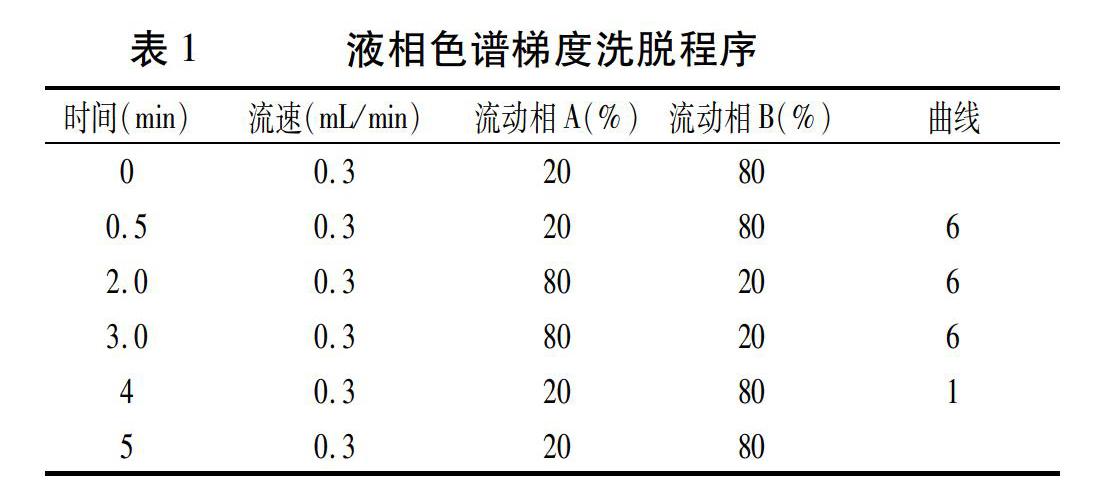
色譜柱:ACQUITY UPLC⑧BEH C18色譜柱,50 mm x2.1mm×1.7μm;流動(dòng)相A:甲醇(0.1%甲酸);流動(dòng)相B:水溶液(0.1%甲酸);流速:0.3mL/min;進(jìn)樣量:10 μL;柱溫:35℃。
液相色譜梯度洗脫程序見(jiàn)表1。
1.4 質(zhì)譜條件
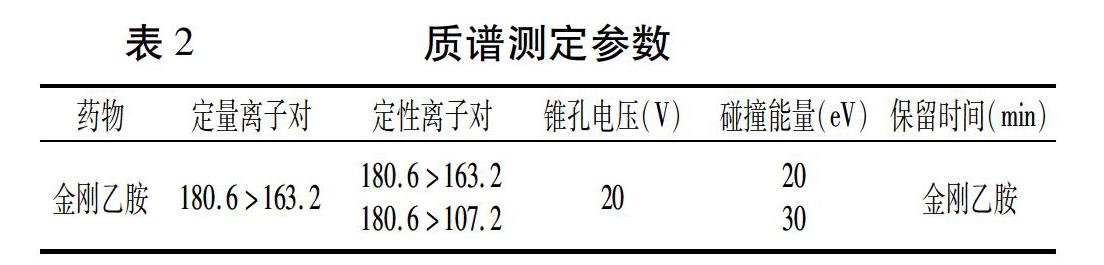
電噴霧離子源;正離子掃描(ESI+);多反應(yīng)監(jiān)測(cè)(MRM);離子源溫度:11O℃;脫溶劑溫度:320℃;脫溶劑氮?dú)饬魉伲?00 L/h;毛細(xì)管電壓:3 kV。
測(cè)試藥物定性、定量離子對(duì)及對(duì)應(yīng)的錐孔電壓和碰撞能量見(jiàn)表2。
1.5 樣品制備
1.5.1 陰性樣品公英青藍(lán)合劑,批號(hào)180216,來(lái)源:煙臺(tái)綠葉動(dòng)物保健有限公司;批號(hào):20180201,來(lái)源:濰坊華英生物科技有限公司。
1.5.2 標(biāo)準(zhǔn)儲(chǔ)備液的制備 精密稱取金剛乙胺標(biāo)準(zhǔn)物質(zhì)10mg(準(zhǔn)確至0.01mg),置100mL容量瓶中,加甲醇溶解并稀釋至刻度,搖勻。-20℃冰箱保存,貯存期3個(gè)月。
1.5.3 樣品溶液的制備 精密量取空白公英青藍(lán)合劑0.2mL,置50mL離心管中,加19.8mL甲醇,超聲處理10 min,5000 r/min離心5 min,過(guò)濾,精密量取續(xù)濾液lmL,置50mL容量瓶中加初始流動(dòng)相稀釋至刻度,搖勻,過(guò)濾,即得空白基質(zhì)溶質(zhì)。待測(cè)樣品以上法處理,作為UPLC -MS/MS供試品溶液。
1.5.4 基質(zhì)匹配標(biāo)準(zhǔn)儲(chǔ)備溶液的制備 精密量取金剛乙胺標(biāo)準(zhǔn)儲(chǔ)備液0.2mL,置20mL容量瓶中,加空白基質(zhì)溶液稀釋至刻度,搖勻,從中精密量取1mL置10 mL容量瓶中,加空白基質(zhì)溶液稀釋至刻度,搖勻,過(guò)濾,即得100ng/mL的基質(zhì)匹配標(biāo)準(zhǔn)儲(chǔ)備溶液。
1.5.5 標(biāo)準(zhǔn)曲線的繪制 上述儲(chǔ)備液(1.5.2)用初始比例流動(dòng)相稀釋,制成0.5、l、2、5、10、20、40、50 ng/mL的溶液,并繪制標(biāo)準(zhǔn)曲線。
1.5.6 陽(yáng)性添加樣品的制備 精密稱取5、10、20mg金剛乙胺分別加入到陰性樣品基質(zhì)200 mL中配制成25、50、100 μg/mL的陽(yáng)性添加樣品,旋渦混勻,每種添加濃度制備3份平行樣,每份樣品至少進(jìn)樣2次,取平均值。
2 結(jié)果與分析
2.1 線性考察
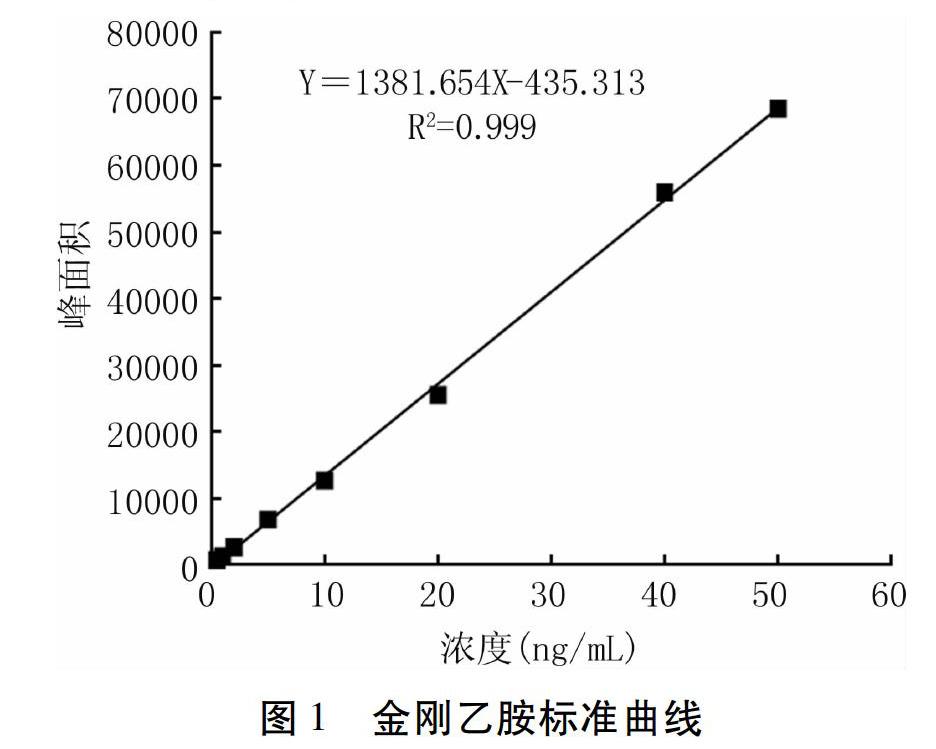
在選定的色譜條件下,使用梯度洗脫的方法,可以有效地分離各離子。取標(biāo)準(zhǔn)曲線0.5、1、2、5、10、20、40、50 ng/mL的溶液,經(jīng)高效液相色譜串聯(lián)質(zhì)譜儀測(cè)定后,以標(biāo)準(zhǔn)溶液中金剛乙胺定量離子峰面積為縱坐標(biāo),標(biāo)準(zhǔn)溶液中相應(yīng)金剛乙胺的濃度為橫坐標(biāo),繪制標(biāo)準(zhǔn)曲線(圖1),在0.5~50ng/mL的范圍內(nèi)標(biāo)準(zhǔn)曲線方程為Y=1381.654X - 435.313,Y為金剛乙胺定量離子峰面積,X(ng/mL)為金剛乙胺的濃度,其相關(guān)系數(shù)R2為0.999,線性良好。
2.2 回收試驗(yàn)
公英青藍(lán)合劑中添加金剛乙胺,液質(zhì)分離良好,無(wú)干擾,公英青藍(lán)合劑中添加濃度5、25、50、100μg/mL,批內(nèi)批間變異系數(shù)均小于10%,陽(yáng)性添加樣品的回收率在80%~120%(表3)。
本方法批內(nèi)相對(duì)標(biāo)準(zhǔn)偏差<10%,批間相對(duì)標(biāo)準(zhǔn)偏差<10%。
對(duì)照溶液、空白樣品及陽(yáng)性添加樣品的液相色譜一串聯(lián)質(zhì)譜譜圖見(jiàn)圖2~圖5。
3 討論與結(jié)論
本研究中,金剛乙胺經(jīng)甲醇超聲提取,稀釋過(guò)濾后,取上清液,用UPLC分離,串聯(lián)質(zhì)譜檢測(cè),外標(biāo)法定量,成功建立了公英青藍(lán)合劑中非法添加的金剛乙胺檢測(cè)方法。該方法經(jīng)過(guò)驗(yàn)證,靈敏度、準(zhǔn)確性、精密度高,檢測(cè)限低,時(shí)間短,速度快,適用于公英青藍(lán)合劑中金剛乙胺非法添加的定性、定量檢測(cè)。
3.1 本試驗(yàn)分別選擇甲醇(0.1%甲酸)和乙腈(0.1%甲酸)為流動(dòng)相A作比較,發(fā)現(xiàn)以甲醇為流動(dòng)相,其特征離子質(zhì)量色譜圖的峰形和分離度比用乙腈做流動(dòng)相好,最終選擇甲醇(0.1%甲酸)為流動(dòng)相A。
3.2 本方法選擇多反應(yīng)監(jiān)測(cè)模式、正離子掃描,并不斷優(yōu)化質(zhì)譜條件:對(duì)離子源溫度、脫溶劑氣流量、錐孔電壓、碰撞能量等參數(shù)進(jìn)行優(yōu)化,使藥物的離子化程度達(dá)到最佳狀態(tài)。計(jì)算結(jié)果采用外標(biāo)法,標(biāo)準(zhǔn)曲線線性良好;樣品經(jīng)過(guò)1000倍的稀釋,上機(jī)檢測(cè)雜峰極微,無(wú)干擾,分離度好。
3.3 本試驗(yàn)針對(duì)公英青藍(lán)合劑中非法添加的金剛乙胺,進(jìn)行了方法學(xué)的考察驗(yàn)證,回收率高,準(zhǔn)確度、精密度良好。下一步需要研究其他中獸藥單方或復(fù)方制劑的金剛乙胺的檢測(cè)方法,為進(jìn)一步制訂國(guó)家標(biāo)準(zhǔn)提供依據(jù),為治理整頓獸藥中添加違禁藥物、嚴(yán)厲打擊違法行為提供檢測(cè)方法。
參考文獻(xiàn):
[1] 中國(guó)獸藥典委員會(huì).獸藥質(zhì)量標(biāo)準(zhǔn)(中藥卷)[M].北京:中國(guó)農(nóng)業(yè)出版社,2017:96 - 97.
[2] 國(guó)家藥典委員會(huì).中華人民共和國(guó)藥典(二部)[M].北京:中國(guó)醫(yī)藥科技出版社,2015:1020.
[3] 齊剛,張秀芹,劉婷,等.超高效液相色譜串聯(lián)質(zhì)譜法檢測(cè)雞蛋中的金剛烷胺和金剛乙胺[J].現(xiàn)代畜牧科技,2017 (1):4-5.7.
[4] 尹暉,孫雷,畢言鋒,等.雞肉和雞蛋中金剛烷胺與金剛乙胺殘留檢測(cè)UPLC - MS/MS法研究[J].中國(guó)獸藥雜志,2014,48(6):32 -35.
[5] 陳慧華,韋敏玨,周煒,等.液相色譜一串聯(lián)質(zhì)譜法測(cè)定動(dòng)物組織中金剛烷胺和金剛乙胺的殘留量[J].質(zhì)譜學(xué)報(bào),2013,34(4):226 -232.
[6] 段科,劉剛,婁喜山,等.超高效液相色譜一串聯(lián)質(zhì)譜法測(cè)定動(dòng)物組織中的金剛烷胺、金剛乙胺和鹽酸美金剛[J].食品安全質(zhì)量檢測(cè)學(xué)報(bào),2018,9(3):652 -658.
[7] 魏秀麗,張傳津,李有志.UPLC - PDA法測(cè)定楊樹(shù)花口服液中非法添加物黃芩苷[J].中國(guó)獸藥雜志,2018,52(1):50 -55.
[8] 魏秀麗,高迎春,楊林,等.超高效液相一串聯(lián)質(zhì)譜法檢測(cè)抗菌藥物中非法添加物利巴韋林[C]∥中國(guó)畜牧獸醫(yī)學(xué)會(huì)動(dòng)物藥品學(xué)分會(huì)第五屆全國(guó)會(huì)員代表大會(huì)暨2016年學(xué)術(shù)年會(huì)論文集.
