連翹敗毒丸高效液相色譜指紋圖譜研究及聚類分析
2019-09-16 02:51:50許翔
中國藥業 2019年18期
許 翔
(中國人民解放軍聯勤保障部隊第九〇〇醫院藥學科,福建 福州 350000)
連翹敗毒丸由連翹、金銀花、黃連、黃芩、黃柏、赤芍等19味中藥材組方,有清熱解毒、散風消腫功效,臨床主要用于治療臟腑積熱與風熱濕毒引起的瘡瘍初起、紅腫疼痛、憎寒發熱、風濕疙瘩、遍身刺癢、大便秘結[1]。本研究中建立了連翹敗毒丸的高效液相色譜(HPLC)指紋圖譜,并結合聚類分析,為其質量控制和快速評價提供借鑒。現報道如下。
1 儀器與試藥
1.1 儀器
1200型高效液相色譜儀(安捷倫科技有限公司,包括在線脫氣機、四元梯度泵、自動進樣器、VWD-G1314C型檢測器、Chemistation數據處理系統);AUW-220D型十萬分之一電子天平(日本島津公司);KQ-250DB型數控超聲波清洗器(昆山超聲波儀器有限公司)。
1.2 試藥
連翹苷對照品(批號為 110821-201816,含量95.1%),連翹酯苷A對照品(批號為111810-201707,含量97.2%),綠原酸對照品(批號為110753-201817,含量96.8%),黃芩苷對照品(批號為110715-201821,含量95.4%),鹽酸小檗堿對照品(批號為110713-201814,含量 86.7%),漢黃芩苷對照品(批號為112002-201702,含量98.5%),芍藥苷對照品(批號為110736-201842,含量97.4%),均購于中國食品藥品檢定研究院;連翹敗毒丸(承德天元藥業股份有限公司,批號分別為 171019,171222,180126,180222,180319,180408,180513,180619,180910,181021,181107,181126,編號為 S1 ~ S12,規格為每袋 9 g);乙腈、甲醇均為色譜純,磷酸為分析純,水為純化水。
2 方法與結果
2.1 色譜條件[2-3]
色譜柱:YMCHydrosphere-C18柱(250mm×4.6mm,5 μm);流動相:乙腈(A)-0.5% 磷酸溶液(B),梯度洗脫 (0~16 min、16%A→25%A,16~30 min、25%A→38%A,30~40 min、38%A→59%A,40~54 min、59%A→80%A,54~62 min、80%A→16%A);流速:0.7 mL/min;檢測波長:328(0~16 m),220 nm(16~40 min),277 nm(40 ~62 min);柱溫:30 ℃;進樣量:10 μL。
2.2 溶液制備
混合對照品溶液:分別稱取連翹苷、連翹酯苷A、綠原酸、黃芩苷、漢黃芩苷、鹽酸小檗堿、芍藥苷的對照品各適量,精密稱定,以甲醇溶解并定容,制成質量濃度分別為 2.245 6,2.001 7,1.113 4,0.582 7,0.511 9,1.201 2,0.894 1 mg/mL的混合對照品溶液,冷藏避光保存。
供試品溶液:取裝量差異項下的內容物,研細,取約2.5 g,精密稱定,置50 mL棕色容量瓶中,加入30 mL甲醇溶液,室溫超聲(功率450 W,頻率40 kHz)處理40 min后取出,放冷,用甲醇定容,搖勻,濾過,即得。
2.3 HPLC指紋圖譜的建立
方法學考察:按標準進行方法學考察,結果精密度、穩定性、重復性試驗結果的RSD均小于2%,表明儀器精密度、方法重復性良好,供試品溶液室溫放置24h內穩定。
指紋圖譜的建立及相似度分析:取12批樣品,依法制備供試品溶液,按2.1項下色譜條件進樣,測定,得HPLC圖譜,導入《中藥色譜指紋圖譜相似度評價系統》(2012A版)軟件,得HPLC指紋圖譜,詳見圖1和圖2。S1~S12號樣品與對照圖譜的相似度分別為0.989,0.971,0.967,0.992,0.909,0.981,0.988,0.974,0.979,0.994,0.993,0.989,均大于 0.9。在 23 個共有峰中,12號峰的面積最大,峰位居中,分離完全,通過與混合對照品圖譜對照,指認出其為連翹苷,設為參照峰(S),計算其他共有峰對12號峰的相對峰面積。詳見表1。
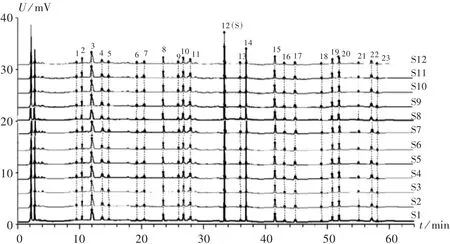
圖1 12批樣品HPLC疊加指紋圖譜
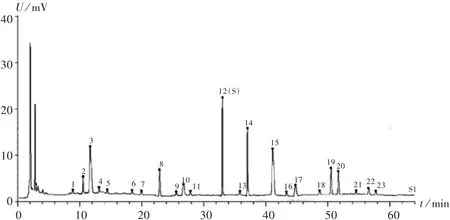
圖2 樣品HPLC對照指紋圖譜
共有峰指認:采用《中藥色譜指紋圖譜相似度評價系統》(2012A版)對12批樣品的HPLC圖譜進行比較分析。結果,可指認23個共有峰中的7個成分,分別為綠原酸(3 號峰)、芍藥苷(8 號峰)、連翹苷(12 號峰)、連翹酯苷A(14號峰)、鹽酸小檗堿(15號峰)、黃芩苷(19號峰)、漢黃芩苷(20號峰)。混合對照品色譜圖見圖3。
2.4 聚類分析[4-5]
將12批樣品中23個共有峰的相對峰面積數據輸入SPSS 22.0統計學軟件,采用組間均聯法,以歐氏距離平方為測度,采用系統聚類分析法進行模式識別分析,結果見圖4。根據聚類分析樹狀圖,當類間距介于15~25時,12批樣品被分成了 3大類:S4、S10、S11為一類,S1、S2、S3、S6、S7、S8、S9、S12 為一類,樣品 S5 單獨為一類。聚類分析結果與相似度結果基本一致。
3 討論
現行質量標準僅對連翹敗毒丸的性狀、大黃的薄層和丸項下的通則檢查做了規定,難以全面評價其質量,文獻報道鮮有對其綜合質量的評價方法[6]。在中醫藥理論中,中成藥的藥效是各味藥發揮協同作用的結果,指紋圖譜可全面控制中藥內在化學成分的整體質量,它是以色譜峰的相對保留時間及相對峰面積來進行定性,以色譜峰面積進行定量,可作為分析中藥內部有效成分的技術[7-8]。
本研究中以12號峰(連翹苷峰)為參照峰,計算得的12批樣品23個共有峰的相對保留時間的RSD為0.05% ~1.10%(文中表略),均較小;而相對峰面積的RSD為 0.06% ~4.72% ,其中 7,18,21,23 號峰大于2.0%,峰面積的差異可能與原料藥材質量有關,不同產地和來源的藥材質量有很大差異,建議藥廠在選藥投料生成前應注意藥材成分含量的檢測。
本研究中曾采用二極管陣列檢測器行全波長掃描,為了實現連翹苷、連翹酯苷A、綠原酸、黃芩苷、漢黃芩苷、鹽酸小檗堿、芍藥苷的同時測定,采用不同時段切換不同檢測波長:0~16 min時,在328 nm波長處測定綠原酸;16~40 min時,在220 nm波長處檢測連翹苷、連翹酯苷A及芍藥苷;40~62 min時,在277 nm波長處檢測黃芩苷、漢黃芩苷、鹽酸小檗堿。同時比較了乙腈和甲醇作為有機相,0.1%磷酸溶液、0.3%磷酸溶液、0.5%磷酸溶液及水作為無機相的檢查效果,結果表明,以乙腈-0.5%磷酸溶液系統得到的指紋圖譜色譜峰數量多、信號強、分離效果好。研究過程中還比較了超聲提取、加熱回流提取及浸漬過夜提取3種提取方法的差異,結果提取率無明顯差異,但超聲提取時間短、操作簡便,故為了節約成本采用超聲提取法。
綜上所述,本研究中建立了連翹敗毒丸HPLC指紋圖譜,實現了對其多指標成分整體、綜合的客觀評價與分析,共識別出23個共有峰,并指認出其中7個指標性成分,能較好地反映出連翹敗毒丸內在質量信息,可為有效控制連翹敗毒丸的質量提供科學依據。
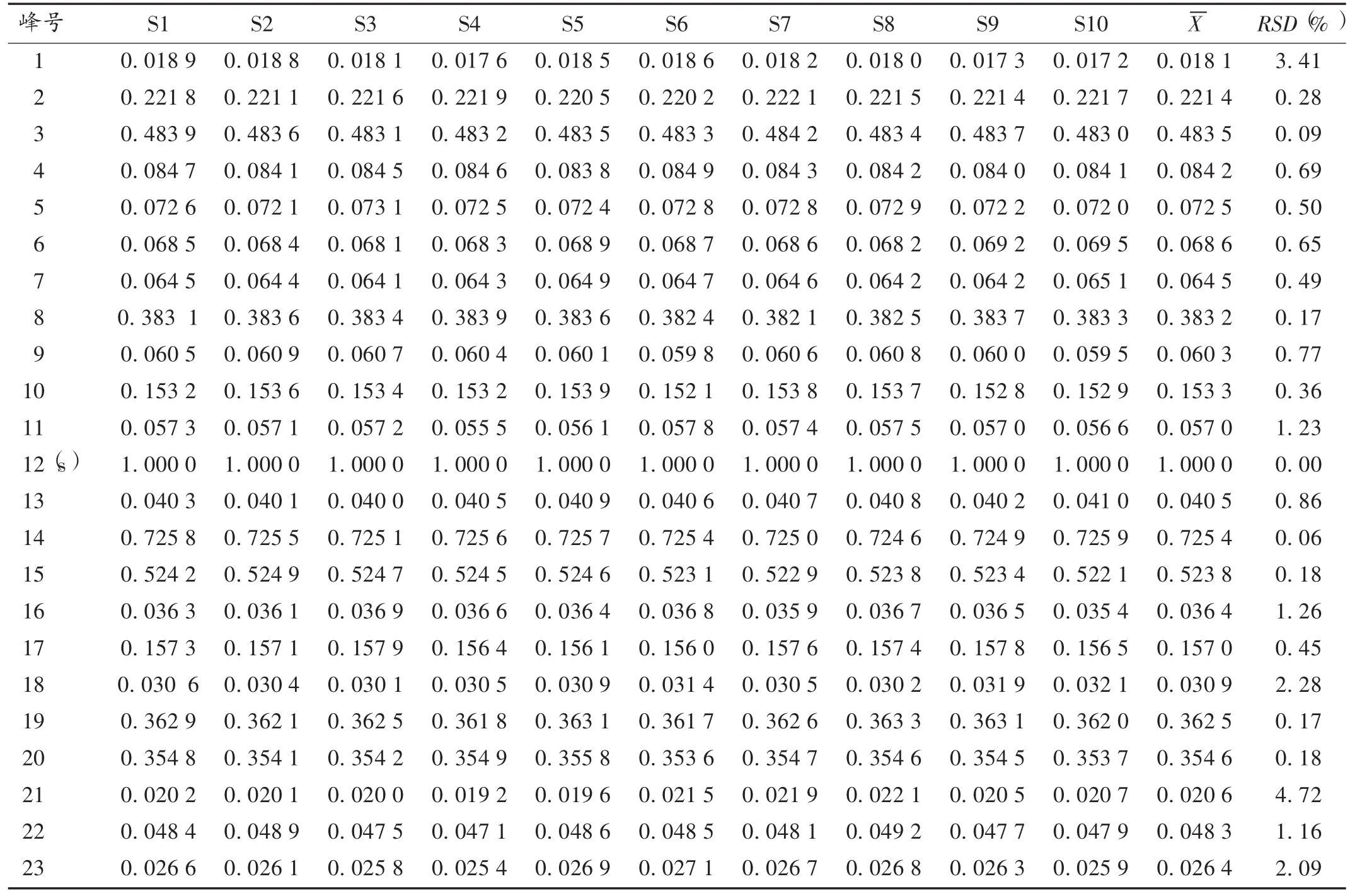
表1 12批藥材樣品HPLC圖譜共有峰的相對峰面積
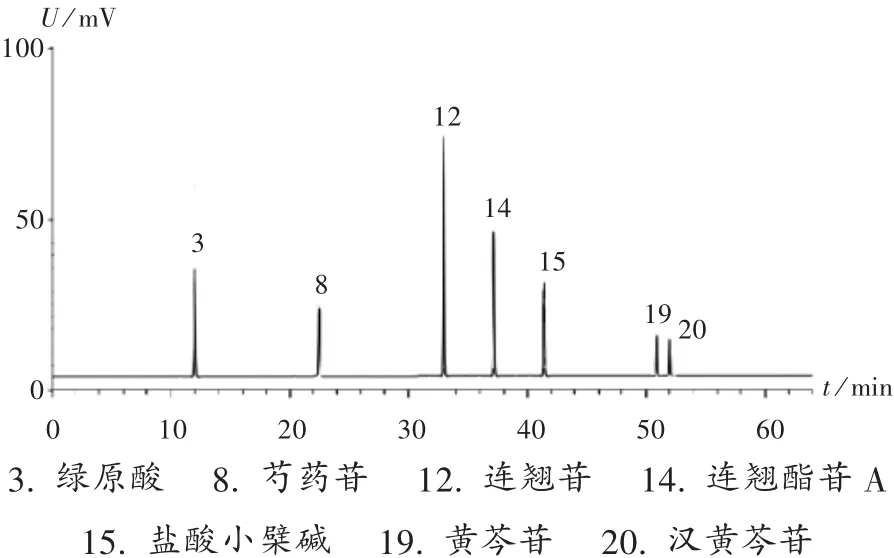
圖3 混合對照品溶液HPLC圖
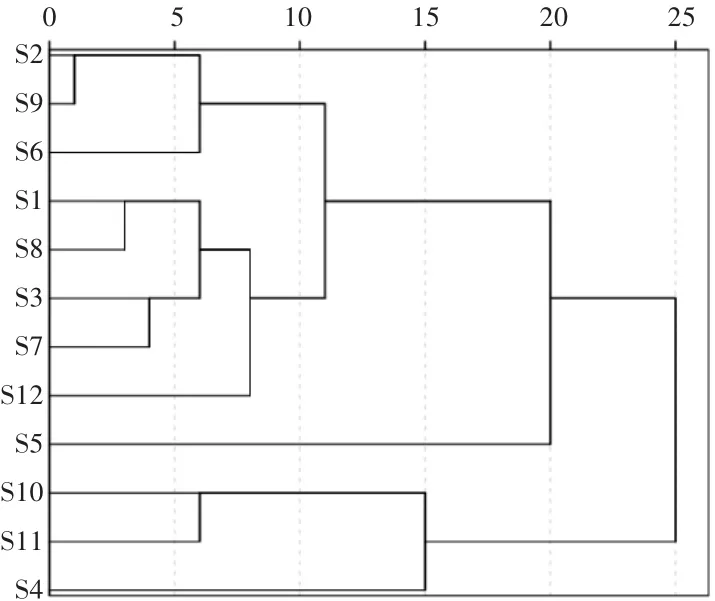
圖4 12批樣品聚類分析樹狀圖
