水楊醛希夫堿橋聯的六核鏑(Ⅲ)配合物的合成、結構及磁性能
2019-09-17 00:59:04馮丹丹楊恩翠趙小軍
天津師范大學學報(自然科學版) 2019年4期
馮丹丹,黃 璐,楊恩翠,趙小軍
(1.天津師范大學化學學院,天津300387;2.天津師范大學無機-有機雜化功能材料化學教育部重點實驗室,天津300387;3.天津師范大學天津市功能分子結構與性能重點實驗室,天津300387)
自從20 世紀90 年代第一個單分子磁體Mn12Ac被發現以來[1],很多基于過渡金屬離子的分子基磁體被合成出來并表現出非常有趣的慢弛豫行為.相對于被廣泛研究的具有較低各向異性能壘(Ueff)和磁阻塞溫度(Tb)的過渡金屬配合物而言[2],由較大磁各向異性和基態自旋的稀土金屬離子形成的稀土金屬配合物開創了分子基磁體的新紀元.稀土單分子磁體表現出的高能壘和高阻塞溫度[3]明顯優于過渡金屬配合物.這些罕見的磁學性能使稀土金屬配合物在量子計算、自旋電子學器件以及高密度信息存儲等方面得以廣泛應用,在化學、物理等多領域都引起了高度關注[4-5].
稀土單分子磁體的各向異性能壘與金屬中心的單離子各向異性和自旋載體間的磁耦合作用緊密相關.因此,研究人員可以通過選擇合適的橋聯配體來調控單分子磁體的磁軸方向,從而改變其慢弛豫磁行為.利用同時含有氮原子和氧原子的希夫堿配體可以有效地將多個稀土金屬橋聯構成多核單分子磁體,這是目前構筑結構新穎的分子基磁體的有效途徑之一[6-7].基于希夫堿配體的單分子磁體的相關研究中,配合物大部分是雙核、三核、四核結構[8-10],其中雙核結構最為常見,而同金屬的六核結構則較為少見.有研究表明,混合配體策略既有利于將金屬中心橋聯形成多核結構,又可以作為封端配體限制配合物的金屬中心的數量,進而實現稀土配合物核數和磁學性能的調控[11].為了合成具有新穎結構和較好磁性能的稀土單分子磁體,本研究選擇水楊醛希夫堿和六氟乙酰丙酮作為混合配體,與稀土Dy(Ⅲ)鹽自組裝反應得到配合物,并對其進行結構表征和性能測試.
1 實驗
1.1 儀器與試劑
儀器: APEX-ⅡCCD X-線單晶衍射儀和D8 ADVANCE 粉末衍射儀,德國Bruker 公司;FT-IR 型紅外光譜儀(KBr 壓片法),美國Nicolet 公司;MPMS3磁性測量儀,美國Quantum Design 公司;CE-440 元素分析儀,美國Leeman Labs 公司.
試劑:六水合硝酸鏑,山東中凱稀土材料有限公司;六氟乙酰丙酮(hfac),北京百靈威科技有限公司.所有試劑均為分析純級.
1.2 配體H2L 與配合物的合成
對文獻[12]中的方法加以改進,合成了配體H2L,合成路線如圖1 所示.用甲醇取代文獻[12]中的乙醇,將水楊醛換成吡啶-2-甲醛,大大提高了配體產率.

圖1 配體H2L 的合成路線Fig.1 Synthesis route of ligand H2L
在室溫攪拌條件下,將H2L(0.2 mmol,48.2 mg)、hfac(0.1 mmol,20.8 mg)、Dy(NO3)3·6H2O(0.2 mmol,91.3 mg)、H2SO3(0.1 mmol,0.8 mg)和三乙胺(0.2 mmol,0.2 mL)緩慢溶解于10.0 mL 的甲醇-乙醇-乙腈的混合溶劑中(體積比為4 ∶2 ∶4).將所得混合物轉移到內襯有聚四氟乙烯的不銹鋼反應釜(23.0 mL)中,并將密閉后的反應釜放入控溫烘箱中.調節控溫程序,將反應釜中的混合物以9.2 ℃/h 的速率由室溫加熱到80 ℃,在此溫度下反應48 h.當烘箱以2.3 ℃/h 的速率降到室溫后,得到適合于單晶X-線衍射的黃色塊狀晶體.經分離后,用甲醇洗滌,置于空氣中晾干,產率為66.7%(基于Dy(Ⅲ)鹽).元素分析以C92H67Dy6F12N21O29S2計,理論值(%):C,34.56;H,2.11;N,9.20.實際值(%):C,34.59;H,2.10;N,9.21.紅外(KBr,cm-1):3405(w),1 656(m),1 597(s),1 499(m),1 438(m),1 469(m),1 382(s),1 252(m),1 149(m),1 052(m),869(m),837(m),765(m).
1.3 配合物的晶體結構測定
在APEX-ⅡCCDX-線單晶衍射儀上,用Mo-Kα射線(λ=0.071 073 nm)在296(2)K 下收集配合物的單晶衍射數據.測量的所有獨立衍射點用于結構分析,用multi-scan 方法通過SADABS 程序[13]進行半經驗吸收校正.使用SAINT 程序[14]對所有的衍射數據進行還原,使用SHELXS-97 和SHELXL-97 程序[15]直接對所有結構進行解析和精修.用全矩陣最小二乘法對除氫以外的所有原子坐標及各向異性溫度因子進行修正,計算氫原子的理論坐標.配合物的CCDC 編號為1864059,晶體學數據和結構精修參數如表1 所示.
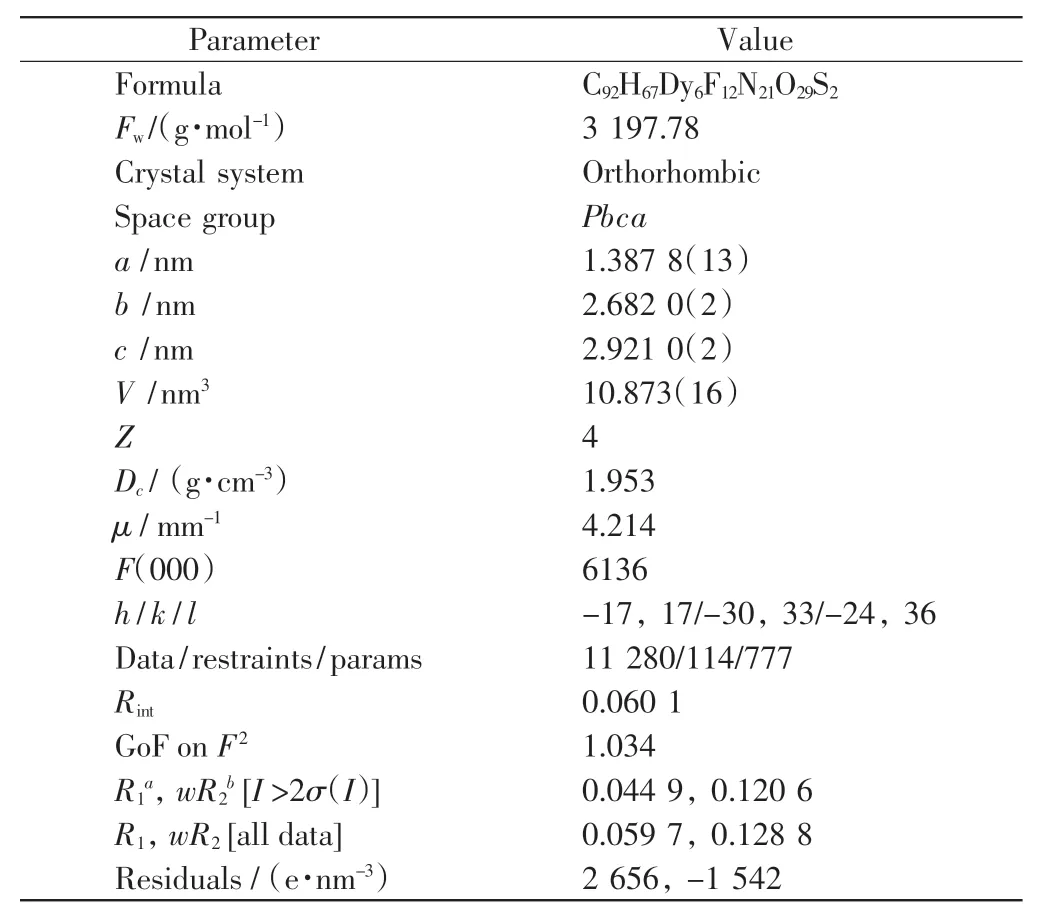
表1 配合物的晶體學數據和精修參數Tab.1 Structural and refinement parameters of the complex
2 結果與分析
2.1 配合物的晶體結構
單晶X-線結構測定結果表明,配合物{[Dy6(HL)2(hfac)2(NO3)2L4(SO3)2]·C2H5OH·CH3CN}結晶于正交晶系Pbca 空間群,呈現出一個中心對稱的籠子狀的六核結構.該配合物的晶體學不對稱單元中包含3 個獨立的Dy(Ⅲ)離子、2 個雙去質子的希夫堿配體、1 個雙去質子的希夫堿配體、1 個六氟乙酰丙酮陰離子、1 個硝酸根陰離子、1 個亞硫酸根陰離子,半個游離的乙醇分子以及半個自由的乙腈分子.配合物的主要鍵長和鍵角數據如表2 和表3 所示,分子結構如圖2 所示.
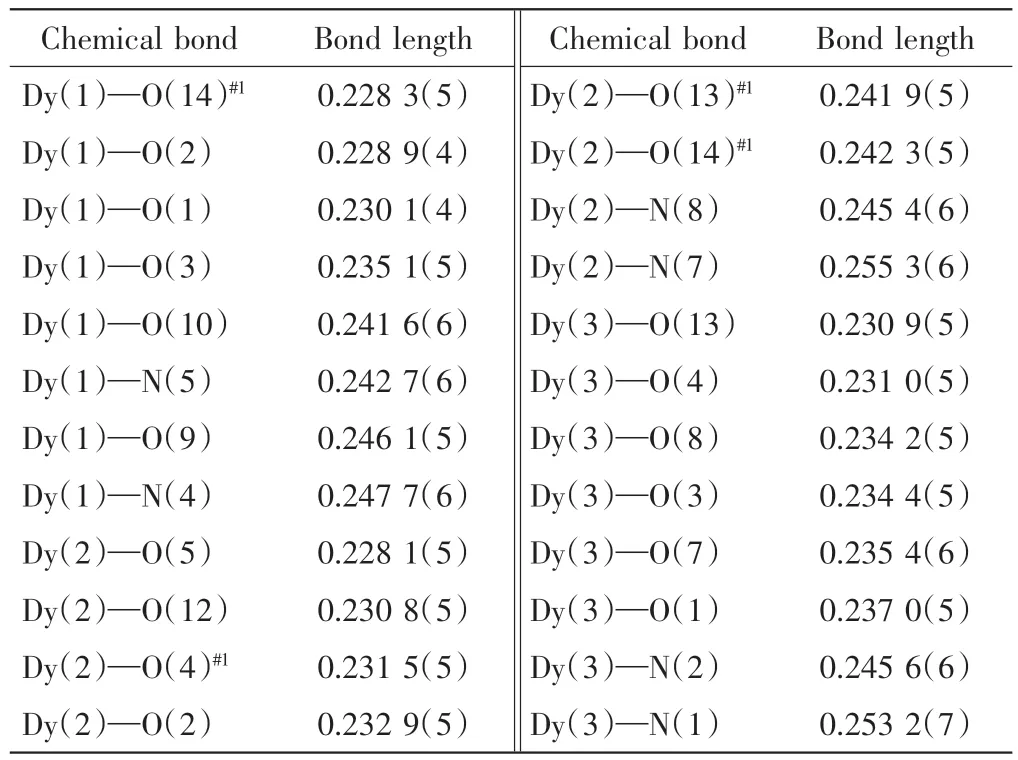
表2 配合物的主要鍵長aTab.2 Selected bond lengths for the complexa nm
由圖2 可知,該配合物中3 個晶體學獨立的Dy1、Dy2 和Dy3 離子均處于八配位的N2O6的配位環境中,呈現扭曲的三角十二面體的配位構型.由SHAPE 軟件計算得到3 個Dy(Ⅲ)離子的一致性因子分別為1.979、2.516 和2.093,表明Dy2 離子的變形程度最大.在Dy1 的配位多面體中,2 個N 原子分別來自于L2-配體的酰肼基(N5)和吡啶基(N4),6 個氧原子分別為來自于L2-配體的羥基氧原子(O2)和2 個酰肼的羰基氧原子(O1 和O3)、NO3-陰離子的2 個螯合氧原子(O9 和O10)和SO32-陰離子的氧原子(O14A).與Dy2配位的2 個N 原子分別來源于單去質子的HL-配體的酰肼基(N8A)和吡啶基(N7A),6 個氧原子分別來自于2 個完全去質子的L2-的羥基氧(O4 和O2A)、1個單去質子的HL-酰肼的羰基氧(O5A)和2 個SO32-陰離子的3 個氧原子(O12A、O13 和O14).與Dy3 配位的2 個N 原子分別來源于完全去質子的L2-配體的酰肼基(N2)和吡啶基(N1),6 個氧原子分別來自于2 個完全去質子的L2-的1 個羥基氧(O4)和2 個酰肼的羰基氧(O1 和O3)、1 個SO32-陰離子的氧原子(O13)和1個hfac-陰離子的2 個氧原子(O7 和O8).由表2可以看出,Dy—O 的鍵長范圍為0.228 1(5)~0.246 1(5)nm,Dy—N 的鍵長范圍為0.242 7(6)~0.255 3(6)nm 之間,與已報道的由希夫堿類配體形成的稀土鏑(Ⅲ)配合物的鍵長范圍一致[16].由表3 可知,Dy(Ⅲ)離子相關的鍵角變化范圍為29.00(11)°~147.14(18)°.

表3 配合物的主要鍵角aTab.3 Selected bond angles for the complexa (°)

圖2 配合物的分子結構Fig.2 Molecular structure of the complex
希夫堿配體在配合物中以單去質子(HL-)和雙去質子(L2-)2 種形式存在.單去質子的HL-配體采取三齒N吡啶基、N酰肼、O酰肼螯合的方式參與Dy(Ⅲ)離子配位多面體的形成,而雙去質子的L2-陰離子則采取μ3-η2: η2: η1: η1的橋聯鍵合模式聚集2 個中心對稱的Dy(Ⅲ)離子.配合物中的SO32-陰離子采取μ4-η2:η2:η1配位模式橋聯4 個Dy(Ⅲ)離子.相比之下,硝酸根和六氟乙酰丙酮陰離子均以雙齒螯合的模式參與Dy(Ⅲ)離子配位多面體的形成.
配合物的核結構如圖3 所示.
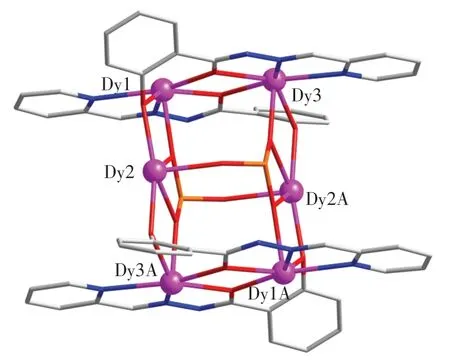
圖3 配合物的核結構Fig.3 Core structure of the complex
由圖3 可以看出,1 對雙去質子的L2-配體通過酰肼氧原子和酚羥基氧原子橋聯Dy1、Dy2 和Dy3 以及Dy1A、Dy2A 和Dy3A,形成2 對中心對稱的{Dy3(L)2}二聚體.這2 對{Dy3(L)2}二聚體進一步通過2 個SO32-陰離子以μ4-η2:η2:η1的鍵合模式聚集在一起,形成封閉籠子狀的六核鏑(Ⅲ)配合物.
2.2 配合物的粉末衍射和紅外光譜分析
配合物的X-線粉末衍射測試在5°≤2θ ≤50°范圍內進行,通過對比理論粉末可知,配合物的實際粉末衍射圖譜與理論模擬圖譜能夠很好地吻合,表明該配合物純度較高.
配合物的紅外光譜在3 405 cm-1處出現的弱峰可歸屬于O—H 的特征伸縮振動,證明了配合物中存在游離的溶劑分子或者是未完全去質子的HL-中的羥基的特征峰[17].在1 656 cm-1和1 469 cm-1處的2 個中等強度的特征吸收峰表明配合物中的金屬中心與六氟乙酰丙酮以烯醇式配位[18].在1 597、1 499 和1 438 cm-1處的中等強度吸收峰歸屬為水楊醛希夫堿配體中酰肼基團的C=O、C=N 的伸縮振動.在1 469 cm-1和1 252 cm-1處的中等強度特征峰為SO32-陰離子的伸縮振動.1 382 cm-1處的強吸收峰和837 cm-1處的中強吸收峰表明存在NO3-陰離子[19].綜上可知,配合物的紅外光譜測定結果與X-線單晶衍射結果一致.
2.3 配合物的磁學性質
在外加1 000 Oe 直流場的情況下測試配合物在2~300 K 范圍內的變溫磁化率曲線,結果如圖4 所示.由圖4 可以看出,300 K 時,配合物的χMT 值為84.61 cm3·K/mol,與6 個獨立的Dy(Ⅲ)離子(S=5/2,g=4/3)的理論值(84.78 cm3·K/mol)一致.隨著溫度降低,配合物的χMT 值逐漸減小.當溫度下降到2.0 K時,測得配合物的χMT 值為41.82 cm3·K/mol,表明配合物中相鄰的Dy(Ⅲ)離子之間存在著弱的反鐵磁相互作用和強的磁各向異性.
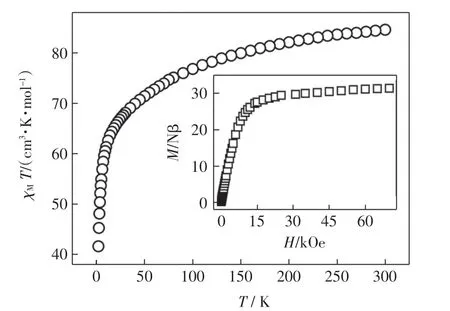
圖4 配合物的變溫磁化率和變場磁化強度曲線Fig.4 Temperature dependence of χMT and field dependence of magnetization(inset)for the complex
在2.0 K 時,測定配合物的變場磁化強度,結果如圖4 插圖所示.隨著外加磁場的不斷增強,配合物的磁化強度不斷增大.當外加磁場為7 T 時,該配合物的磁化強度為31.42 Nβ,遠低于6 個Dy(Ⅲ)離子的理論飽和值(60.0 Nβ),表明該配合物中存在較強的磁各向異性.
為進一步探究配合物的磁化動力學特征,對配合物進行零場和加場條件下的交流磁化率的表征.在零直流場以及2.5 Oe 振蕩交流場下,配合物的變溫交流磁化率的測試在0.1~1 000 Hz 范圍內進行,結果如圖5(a)所示.隨著溫度降低,交流磁化率的實部值不斷增加,且到達9.0 K 時,不同頻率的實部上升趨勢的大小發生改變;而在30.0~12.0 K 范圍內,虛部值隨溫度降低沒有發生明顯變化,低于12.0 K 時,500 Hz 和1 000 Hz 的虛部明顯上升,1 Hz 的虛部則依然無明顯上升.由于不同頻率的實部、虛部曲線均不重合,因此,配合物在零場下表現為頻率依賴,呈現磁矩慢弛豫行為.在低溫時,虛部曲線一直呈現上升趨勢且未出現峰值,這是由量子隧穿效應造成的.
當外加直流場強度增加到5.0 kOe 時,交流磁化率的虛部在3~10 K 溫度范圍內出現明顯的峰值,且隨溫度的不斷升高,交流磁化率虛部的峰值在高溫區隨頻率移動.這說明加場條件有效抑制了量子隧穿.利用公式ln(χ″/χ′)=ln(ωτo)+ΔE/kBT 對各向異性能壘(ΔE/kB)和指前因子(τo)進行估算(見圖5(b)插圖),得到ΔE/kB≈11.83 K 以及τo≈5.29×10-5s.

圖5 配合物的實部和虛部交流磁化率對頻率曲線(插圖:配合物的ln(χ″/χ′)對1/T 曲線)Fig.5 Frequency dependence of the in-phase(χ′)and out-of-phase(χ″)ac susceptibilities for the complex(Insert:Plot of ln(χ″/χ′)versus 1/T for the complex)
3 結論
本文在亞硫酸存在條件下,通過溶劑熱方法制備了一個以水楊醛希夫堿和六氟乙酰丙酮為混合配體的稀土鏑(Ⅲ)配合物.結構測定結果表明,水楊醛希夫堿配體和亞硫酸根陰離子聚集離散的鏑(Ⅲ)離子形成了一個新穎的封閉籠子狀的六核鏑(Ⅲ)配合物.該配合物中相鄰的鏑(Ⅲ)離子間存在著弱的反鐵磁相互作用,在零場下呈現磁矩的慢弛豫行為,在外加5.0 kOe 直流場下表現為單分子磁體行為.
