混播比例和刈割茬次對多花黑麥草+箭筈豌豆混播草地土壤細菌群落的影響
2019-11-07 11:54:54茍文龍董臣飛李達旭白史且師尚禮
草地學報 2019年5期
茍文龍, 李 平, 董臣飛, 李達旭, 白史且, 師尚禮*
(1.甘肅農業大學草業學院, 甘肅 蘭州 730070; 2. 四川省草原科學研究院, 四川 成都 611731;3. 江蘇省農業科學院畜牧研究所, 江蘇 南京 210014)
土壤微生物是土壤生態系統中最為活躍的部分,參與90%左右的土壤反應過程,在土壤物質循環、能量流動及維持土壤生態系統多樣性與穩定性方面發揮至關重要的作用[1]。其中土壤細菌種類繁多,數量最多,約占土壤微生物總量的70%~90%[2],它直接或間接參與土壤生物化學循環,如分解與合成有機物質,土壤結構及腐殖質的形成等[3]。近年來人們開始關注和認識土壤微生物的生態功能,利用土壤微生物結構來衡量土壤健康質量的優劣。土壤細菌群落多樣性是評價土壤質量的一項重要指標。土壤中細菌多樣性越高,越有利于土壤恢復力和抗壓力的提高[4]。植被類型和多樣性、土壤水分、pH值、土壤類型及其理化性狀、土地利用方式均顯著影響微生物群落結構[5],如Lauber等認為pH值是決定土壤微生物群落結構的主要因素[6]。
禾豆混播可以提高單位面積的產草量和蛋白質產量,提高土壤肥力,減少工業氮肥的施用,降低生產成本,減少環境污染,不僅能增加地上部的生物多樣性,也可以增加地下部的土壤微生物多樣性[7-8]。目前禾豆混播的研究主要集中在產量和品質[9]、競爭共存[10]、土壤養分[11]、根系形態[12]、施肥管理[13]、氮素固定與轉移[14]等方面。關于禾豆混播草地土壤細菌群落結構的研究和報導較少,其中陸炳章研究多花黑麥草(LoliummultiflorumLamk.)與光葉紫花苕(ViciavillosaRoth)、箭筈豌豆(ViciasativaL.)的混播結果表明,混播區根際細菌、放線菌、真菌、固氮菌、纖維分解菌等均顯著高于單播和休閑地[15]。王旭等研究燕麥(Avena sativa L.)和箭筈豌豆間行混播收獲期根際土壤細菌數量是單作燕麥的1.1倍左右[16]。而混播比例和刈割茬次對禾豆混播草地土壤群落結構的影響方面的研究就更少。因此,本研究以多花黑麥草和箭筈豌豆混播草地土壤為研究對象,通過直接提取土壤細菌基因組總DNA,采用16S rDNA基因的通用引物PCR擴增及Illumina平臺Miseq高通量測序技術對多花黑麥草和箭筈豌豆混播草地土壤中細菌群落結構與組成、物種豐富度和物種多樣性進行研究,探討混播比例和刈割茬次對多花黑麥草和箭筈豌豆混播草地土壤細菌群落的影響,為深入了解禾豆混播草地土壤細菌群落結構與組成變化、調節土壤環境質量和改善土壤管理提供理論依據。
1 材料與方法
1.1 樣地描述與試驗設計
1.1.1試驗區自然概況 試驗地位于四川省草原科學研究院大邑縣韓場基地(103°45′ E,30°25′ N,海拔475 m)。試驗區域屬大陸性熱帶濕潤季風氣候,年平均氣溫15℃,最熱月7月平均氣溫26.1℃,最冷月1月平均氣溫5.5℃,極端最低氣溫-4.8℃,極端最高氣溫35.1℃,年降水量1 300 mm。土壤為黃粘土,pH 6.74,有機質32.2 mg·kg-1,堿解氮185 mg·kg-1,有效磷41.8 mg·kg-1,速效鉀127.3 mg·kg-1。全年日照時數1 033.8 h,年平均無霜期284 d。
1.1.2供試材料 長江2號多花黑麥草(LoliummultiflorumLamk‘Changjiang No. 2’),純凈度98%,發芽率92%,由四川農業大學草學系提供;川北箭筈豌豆(ViciasativaL‘Chuanbei’),純凈度95%,發芽率94.5%,由四川省農科院土肥所提供。
1.1.3試驗設計 試驗采用完全隨機區組設計,共設5個處理,即100%多花黑麥草(R1)、75%多花黑麥草+25%箭筈豌豆(R2)、50%多花黑麥草+50%箭筈豌豆(R3)、25%多花黑麥草+75%箭筈豌豆(R4)和100%箭筈豌豆(R5),每個處理3個重復。小區面積15 m2(3 m×5 m),區組間隔0.8 m,小區間隔1 m。試驗于2016年9月28日播種,采用撒播,撒播時,多花黑麥草和箭筈豌豆分開撒播。多花黑麥草單播播種量22.5 kg·hm-2,箭筈豌豆單播播種量75 kg·hm-2,混播組合中每個草種播種量是用混播組合中該草種的混播比例與單播播種量的乘積來表示。試驗水肥采用統一管理,分別在翌年的1月4日(H1,多花黑麥草分蘗期/箭筈豌豆分枝期)、3月14日(H2,多花黑麥草拔節期/箭筈豌豆分枝期)、4月21日(H3,多花黑麥草孕穗期/箭筈豌豆現蕾期)、5月19 日(H4,多花黑麥草乳熟期/箭筈豌豆盛花期)4個時期進行刈割。
1.2 土壤取樣和前期處理
每次刈割后,各小區以S形取樣法在株叢附近用土鉆采集0~20 cm土樣,所有土壤樣品采用北京富益聯有限公司的便攜式冰箱(FYL-YS-60L)帶回實驗室,儲存在—80℃超低溫冰箱后進行DNA提取和細菌群落分析。
1.3 DNA提取和 PCR擴增
參照Zeng等的方法進行DNA提取和PCR擴增[17]。使用CTAB / SDS方法提取土壤樣品的總DNA。在1%瓊脂糖凝膠上檢測DNA濃度和純度。使用無菌水將DNA稀釋至1 ng·μL-1。選擇引物515F(5′-CCTACGGGAGGCAGCAG-3)和806R(5′-GGA CTA CHV GGG TWT CTA AT-3′)擴增16S rRNA基因的V4高變區[18]。所有PCR反應均用Phusion?High-Fidelity PCR Master Mix(New England Biolabs)進行。將相同體積的1×上樣緩沖液(含有SYB綠)與PCR產物混合,并在2%瓊脂糖凝膠上進行電泳檢測。選擇具有400 bp和450 bp之間的明亮主條帶的樣品用于進一步實驗。將PCR產物以等密度比混合,并用Qiagen Gel Extraction Kit(Qiagen,Germany)純化。
1.4 文庫建立及測序
使用TruSeq?DNA PCR-Free樣品制備試劑盒(Illumina,USA)按照制造商的推薦產生測序文庫,并添加指數代碼。使用Qubit@2.0熒光計(Thermo Scientific)和Agilent Bioanalyzer 2100系統評估文庫質量。最后,將文庫在Illumina HiSeq2500平臺上測序,并產生250 bp的配對末端讀取數(reads)。
使用FLASH(V1.2.7)合并成對末端讀取數(reads)[19],然后在特定過濾條件下進行,以根據QIIME(V1.7.0)的質量控制過程獲得高質量的有效序列(clean tags)[20]。使用UCHIME算法將序列(tags)與Gold數據庫進行比較,以檢測嵌合體(chimera)序列,然后去掉嵌合體序列[21]。
序列分析由Uparse軟件(Uparse v7.0.1001)完成[22]。具有≥97%相似性的序列被歸類為相同的操作分類單元(OTU,Operational Taxonomic Unit)。對于每個代表性序列,依據GreenGene數據庫[23],采用RDP 3算法(version 2.2)進行分類信息注釋[24]。為了研究不同OTU的系統發育關系,以及不同樣品(組)中優勢種的差異,使用MUSCLE軟件(版本3.8.31)進行多序列比對。
1.5 數據分析
用QIIME(Version 1.7.0)計算alpha多樣性并用R軟件(Version 2.15.3)顯示。PC軟件分析由R軟件(Version 2.15.3)中的WGCNA軟件包,stat軟件包和ggplot2軟件包顯示。Beta多樣性由QIIME軟件(Version 1.7.0)計算。PC軟件分析由R軟件(Version 2.15.3)中的WGCNA軟件包,stat軟件包和ggplot2軟件包顯示。采用方差分析(ANOVA)評估土壤性質和細菌群落指數變化程度。
2 結果與分析
2.1 多花黑麥草和箭筈豌豆混播草地土壤細菌多樣性
土壤樣本細菌alpha多樣性統計如表1所示。通過數據庫比對注釋,得到97 021個OTU,這些OTU具有>96%的序列相似性,R2H3具有最高的OTU(5 456),R4H2具有最低的OTU(3 846)。土壤樣品中細菌Shannon指數和Chao 1指數分別為8.82~10.56和6 697.67~7 963.69,表明禾豆混播草地土壤具有較高的細菌多樣性。混播比例和刈割茬次對一年生禾豆混播草地土壤細菌香農指數和覆蓋率無交互效應,混播比例對土壤alpha多樣性指數無顯著性影響,刈割茬次對土壤細菌OTU和Chao 1指數影響效果顯著(P<0.05)。隨著刈割茬次的增加,多花黑麥草單播R1的土壤細菌OTU和Chao 1指數呈增加趨勢(P<0.05);混播R2的土壤細菌OTU、Shannon指數和系統發育多樣性指數呈先降低后增加趨勢(P<0.05);混播R3的土壤細菌OTU,Chao 1指數和系統發育多樣性指數呈“S”型變化,Shannon指數呈線性增加(P<0.05);混播R4和箭筈豌豆單播R5的土壤細菌OTU,Shannon指數,Chao1指數和系統發育多樣性指數先降低后增加(P<0.05)。
2.2 多花黑麥草和箭筈豌豆混播草地土壤細菌群落組成
如圖1所示,第1茬(H1),R1,R2,R3,R4和R5特有OTU分別為824,620,548,597和975,共有OTU為1 909,第2茬(H2),R1,R2,R3,R4和R5特有OTU分別為780,585,568,482和745,共有OTU為1 746;第3茬(H3),R1,R2,R3,R4和R5特有OTU分別為618,610,864,553和523,共有OTU為2 602;第4茬(H4),R1,R2,R3,R4和R5特有OTU分別為619,579,590,635和566,共有OTU為2 599;不同混播比例之間,R1,R2,R3,R4和R5特有OTU分別為949,732,791,708和762,共有的OTU為5 055;不同刈割茬次之間,H1,H2,H3和H4特有OTU分別為1 236,783,1 913和1 569,共有的OTU為5 073;隨著禾豆混播比例或刈割茬次的增加,特有的OTU呈“Z”型變化;R5H1具有最多的OTU(975),而R4H2具有最少的OTU(482)。

表1 土壤樣本細菌Alpha多樣性統計
注:H1:第1茬刈割,H2:第2茬刈割,H3:第3茬刈割,H4:第4茬刈割。下同
Note:H1:First cut,H2:Second cut,H3:Third cut,H4:Forth cut. The same as below

圖1 多花黑麥草和箭筈豌豆混播草地土壤細菌OTU韋恩圖
多花黑麥草和箭筈豌豆混播草地土壤細菌在門、目和屬分類水平上的相對豐富度如圖2所示。在門水平上的主要群體是變形菌門(Proteobacteria)、放線菌門(Actinobacteria)、厚壁菌門(Firmicutes)、擬桿菌門(Bacteroidetes)、酸桿菌門(Acidobacteria)、芽單胞菌門(Gemmatimonadetes)、硝化螺旋菌門(Nitrospirae)、浮霉菌門(Planctomycetes)、綠彎菌門(Chloroflexi)和疣微菌門(Verrucomicrobia),其中變形菌門、酸桿菌門和放線菌門是土壤的優勢細菌門,占整個細菌類群豐富度的59.1%~69.8%。H3土壤樣品中變形菌門、酸桿菌門和擬桿菌門的豐富度最高(0.34,0.17,0.09),放線菌門豐富度最低(0.11),而H4土壤樣品具有最高豐富度的硝化螺旋菌(0.05);混播比例對門水平土壤細菌豐富度影響不顯著。在目水平上,R3H1具有較高的假單胞菌目(Pseudomonadales),相對豐富度為0.17;鞘脂桿菌目(Sphingobacteriales)隨著刈割茬次增加,豐富度先增加后降低;紅螺菌目(Rhodospirillales)、變形細菌目(Myxococcales)及Gaiellales豐富度隨著刈割茬次增加,相對豐富度先降低后增加。在屬水平上,芽單胞菌屬(Gemmatimonas)和Gaiella隨著禾豆混播中豆科牧草比例的增加,相對豐富度先增加后降低;隨著刈割茬次的增加,芽單胞菌屬和棘刺桿菌屬(Sphingobacterium)呈降低趨勢。
不同混播比例之間未發現具有統計學差異的細菌,不同茬次之間具有統計學差異的土壤細菌(圖3)。H1具有顯著性差異的土壤細菌為γ-變形菌綱(Gammaprobacteria)、假單胞菌目、假單胞菌科(Pseudomonadaceae)、假單胞菌屬(Pseudomonas)和丙酸桿菌目(Propionibacteriales)。H2具有顯著性差異的土壤細菌為嗜熱油菌綱(Thermoleophilia)、紅桿菌目(Solirubrobacterales)、Gaiellales、芽單胞菌目(Gemmatimonadales)、芽單胞菌科(Gemmatimonadaceae)、未分類的芽單胞菌門(undentified-Gemmatimor)、浮霉菌門(Planctomycetales)、浮霉菌綱(Planctomycetacia)和浮霉菌科(Planctomycetaceae)。H3具有顯著性差異的土壤細菌為β-變形菌綱(Betaprotebacteria)、鞘脂桿菌目(Sphingobacteriales)、鞘脂桿菌綱(Sphingobacteria)、Subgroup-6和噬幾丁質菌科(Chitinophagaceae)。H4具有顯著性差異的土壤細菌為未分類的放線菌綱(unidentified-Actinobacteria)、α-變形菌綱(Alphaproteobacteria)、根瘤菌目(Rhizobiales)和紅螺菌目。


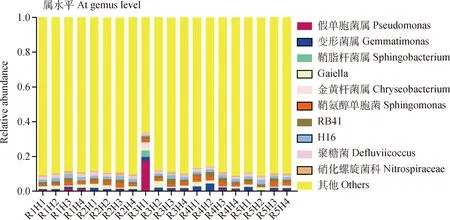
圖2 多花黑麥草和箭筈豌豆混播草地土壤細菌在門、目和屬分類水平上的相對豐富度
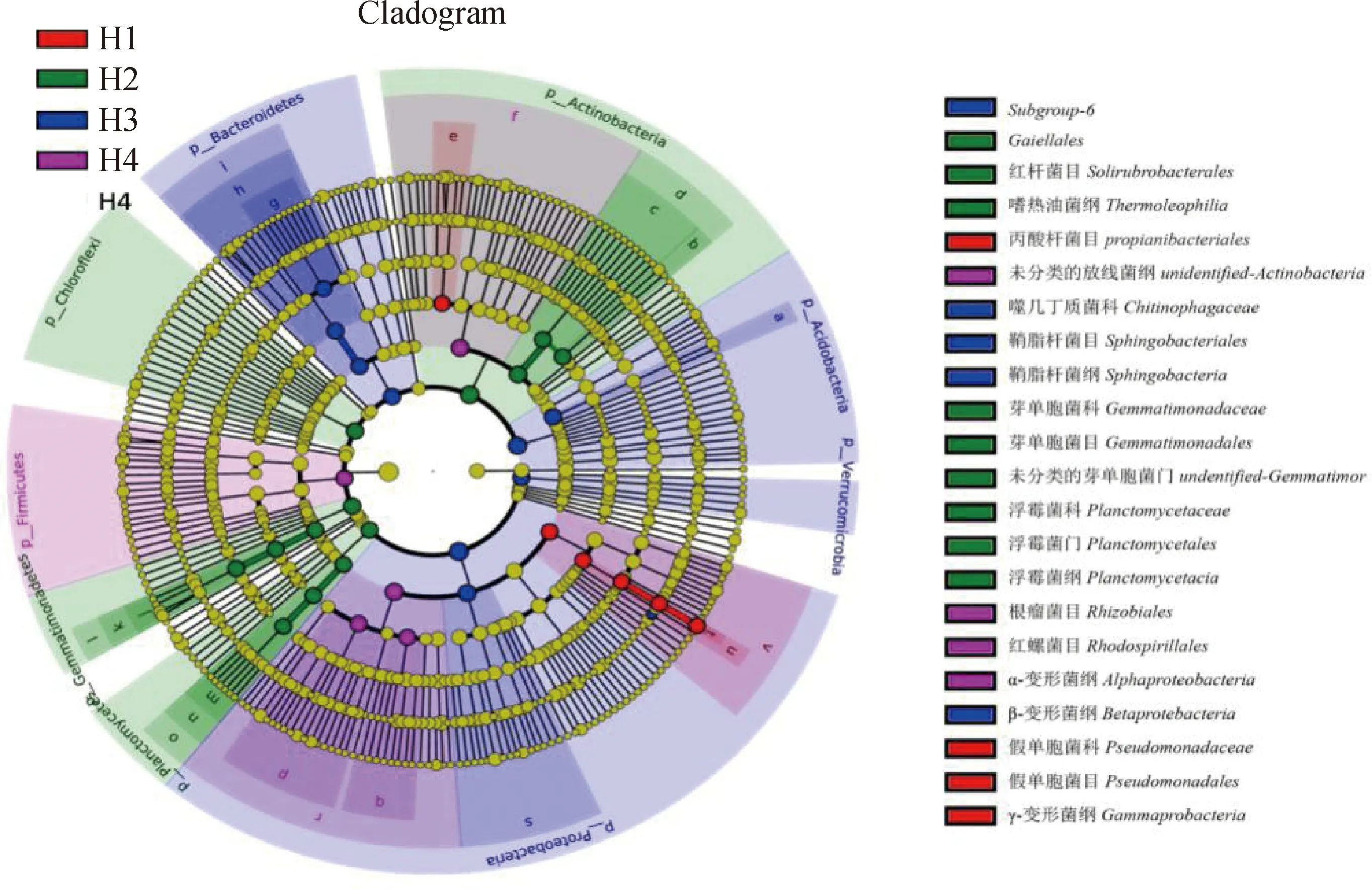
圖3 多花黑麥草和箭筈豌豆混播草地不同刈割茬次之間的LEfSe分析
PCoA分析表明,在X軸和Y軸上分別能夠解釋不同樣品處理之間差異的25.25%和17.61%(圖4)。NMDS分析時,stress為0.127,能夠有效解釋不通樣品的變異程度。PCoA和NMDS分析表明H3的細菌群落顯著不同于H1,H2和H4。通過Annosim,MRPP和Adonis分析時(表2),不同刈割茬次之間細菌多樣性存在顯著差異(P<0.05)。
3 討論
南方是中國主要農業區,傳統種植業由于工業氮肥的持續使用,導致大氣氮沉積,土壤酸化和土壤微生物多樣性降低。如中國南方化肥的大量使用加劇了土壤酸化(從1982年的6.5降至2012年的6.19)和耕地退化,導致土壤生態功能下降[25]。土壤微生物是土壤有機物循環的基礎,其對生存環境改變的相應較迅速,能夠有效及時地反映土壤環境的變化,是表征土壤健康的生物標準[26]。土壤類型、土壤理化性狀、地上植被、季節更替、田間管理等影響土壤微生物的群落組成和結構。其中,豆科牧草的加入有可能提高一年生禾草生產系統的粗蛋白來源和維持土壤健康[27-28]。因此,探索四川農區一年生禾豆混播草地土壤的細菌群落特征,能夠更好地了解這些細菌群落結構短期變化。
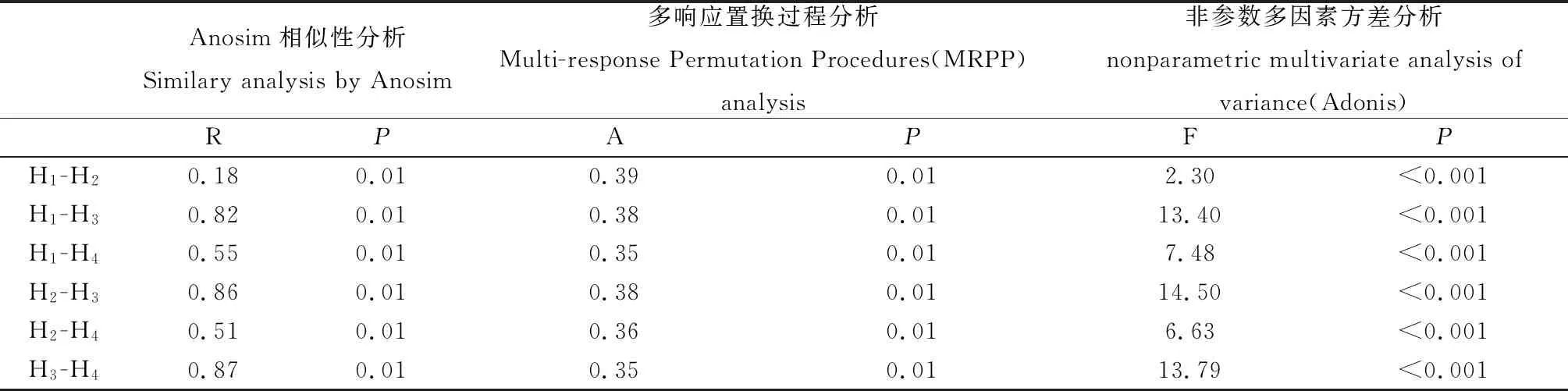
表2 采用3種非參數多變量方法(Anosim、MRPP和Adonis)分析不同刈割茬次對多花黑麥草和箭筈豌豆混播草地土壤細菌群落結構的影響

圖4 多花黑麥草和箭筈豌豆混播草地土壤細菌群落結構
本研究主要利用高通量測序方法探討了混播比例與刈割茬次對多花黑麥草和箭筈豌豆混播草地土壤細菌多樣性的影響,其彌補了傳統培養方法造成的信息丟失缺陷,可以更全面的反映真實環境中土壤微生物的群落結構和多樣性差異。本研究中,總共獲得97 021個OTU,測序的覆蓋率大于96%,表明以上測序量能夠很好地反映各個樣品中細菌群落的結構和種類。所測定序列中主要的門包括變形菌門、放線菌門、厚壁菌門、擬桿菌門、酸桿菌門、芽單胞菌門、硝化螺旋菌門、浮霉菌門、綠彎菌門和疣微菌門主要分布于土壤中,總相對豐富度>0.93。其中三個最豐富的土壤細菌群落是變形菌門(0.29~0.54)、放線菌門(0.08~0.36)和酸桿菌(0.08~0.18),是多花黑麥草和箭筈豌豆混播草地主要的細菌類群。這與多數有關農田土壤微生物群落組成所得研究結果是基本一致的。Ding等曾報道,變形菌門是農田土壤中的主要微生物(相對豐富度0.29~0.33),其次是酸桿菌門(相對豐富度0.12~0.16)和放線菌門(相對豐富度0.09~0.11)[29]。此外,國內關于農田土壤細菌多樣性的測序研究也表明,土壤中優勢細菌門為變形菌門、放線菌門、酸桿菌門、厚壁菌門和芽單胞菌門[5]。
牧草栽培管理措施是影響土壤養分循環和改變微生物群落的重要因素[30]。禾豆混播,地上和地下空間分布格局合理,土壤理化性質改善,明顯地促進了微生物活動,加速了有機物分解和養分積累[31]。植物群落多樣性越豐富,凋落物和根系分泌物組成就越豐富,土壤微生物豐富度和多樣性也就越高[32]。而本研究中,禾豆混播比例對土壤細菌多樣性指標沒有顯著性影響。這可能是因為混播中多花黑麥草生長旺盛、競爭力強,箭筈豌豆生長較弱、耐刈性較差,無論混播比例大小,多花黑麥草始終在混播中占主導地位,使得混播比例對土壤細菌多樣性在短期內未產生較大影響。這與Zhao等關于土壤微生物群落結構在豆科牧草單播、禾本科牧草單播及禾豆混播之間無顯著差異的結果基本是一致的[33]。在溫室環境中進行的生物修復實驗也證實此類現象[34]。因此,多花黑麥草和箭筈豌豆混播草地栽培管理過程中,箭筈豌豆增加了土壤微生物群落中特定物種豐富度。
本研究中,刈割茬次對土壤細菌多樣性有顯著性影響(P<0.05),即隨著刈割茬次的增加,Shannon指數呈線性增加。這說明了刈割引起牧草地上部發生變化后,牧草地上部的光合作用、呼吸作用、蒸騰作用以及光合作用產物的分配與轉運以及根系的分泌等生理生態活動會發生相應的變化[35],這些變化又會通過根系對土壤微生態環境產生一定的影響[36]。賽吉日呼分析了單播苜蓿、單播老芒麥、單播飼用燕麥和混播播種后土壤微生物多樣性,結果表明,隨著牧草生育期的推遲,細菌Alpha多樣性指數呈上升趨勢[37]。PCoA分析表明來自H3的土壤傾向于聚集在一起,表現出具有高度的相似性。由于多花黑麥草和箭筈豌豆混播第3茬刈割在4茬刈割中的單茬地上生物量最大[38],推測認為地上生物量大,根系代謝作用加強,導致凋落物和根系分泌物多,在一定程度上改善了土壤微環境,使得細菌群落結構發生改變。此外,包括變形菌門、放線菌門和酸桿菌門在內的最主要的門在不同刈割時期間發生顯著變化。δ-變形菌綱(Deltaproteobacteria)相對豐富度在H2樣品中發生顯著變化,而α-變形菌綱、β-變形菌綱和γ-變形菌綱相對豐富度在H3樣品中發生顯著變化。這些變化可能與植株長勢的差異、根系活力的差異以及土壤本身和土壤中微生物基數的差異,引起土壤細菌相組成發生顯著變化有關。
4 結論
在門分類水平上,變形菌門、酸桿菌門和放線菌門是多花黑麥草和箭筈豌豆混播草地土壤的優勢細菌門,占整個細菌類群豐富度的59.1%~69.8%,其中組合R3H4(50%多花黑麥草+50%箭筈豌豆混播第4茬刈割)的豐富度最大為69.8%。
在多花黑麥草和箭筈豌豆混播草地中,混播比例對土壤細菌多樣性無顯著性影響,刈割茬次顯著影響土壤細菌OTU和Chao 1指數,引起土壤細菌群落結構和組成發生改變。
