硫酸氫酯類藥劑浮選銅的量子化學研究
2019-12-06 09:31:36慕紅梅馬海濤成莉燕張建輝李文雅
原子與分子物理學報 2019年6期
慕紅梅, 馬海濤, 成莉燕, 張建輝, 李文雅
(1.蘭州資源環境職業技術學院 環境與化工系, 蘭州 730021; 2.蘭州市第三十四中學 物理教研室, 蘭州 730050)
1 引 言
礦產資源是人類賴以發展的物質基礎. 我國雖礦產資源豐富, 但隨著開發規模擴大, 形勢越來越嚴峻, 而我國的經濟發展對礦物材料仍持有高位需求, 解決經濟發展與礦產資源緊缺之間的矛盾已成為一項新的挑戰[1]. 礦物浮選法是極為重要的礦產資源開發手段, 浮選捕收劑的品種和質量直接關系到浮選工藝發展效果的優劣, 而浮選藥劑大都對環境有不同程度的污染, 大規模試驗并不科學. 因此, 針對特定的浮選任務, 設計出新型高效、廉價、環保的浮選藥劑, 對我國礦物的綜合回收利用以及生態環境安全具有重要意義. 開發新型浮選藥劑的方法有物理化學方法、拓撲學方法和分子模擬方法[2,3]. 近年來發展比較迅速的分子模擬方法包括量子化學方法和分子力學動力學方法, 采用分子模擬技術可以降低浮選新藥劑開發的經濟成本, 保護環境, 提高設計效率, 這對于現存的復雜難選礦物的開發利用具有重大的理論指導和實際應用價值.
近年來, 國內外運用基于密度泛函理論的量子化學方法研究設計浮選藥劑主要集中在黃鐵礦和黃銅礦的分子結構[4,5]、黃藥[6,7]、胺類[8]等浮選藥劑. 未有關于硫酸氫酯類浮選藥劑的相關研究報道. 本研究選用15~21個碳原子的硫酸氫酯, 研究其幾何參數、電荷密度分布及與銅離子吸附后能量變化. 通過以上研究能為浮選藥劑設計提供理論依據和設計方法.
2 計算方法
采用密度泛函(DFT)B3LYP方法[9,10], 6-311+G(d, p)基組[11], 對碳原子數為15~21的硫酸氫酯進行參數優化, 得到各物質穩定結構、 能量和電荷分布. 進一步計算了各物質與銅離子作用后的穩定結構和吸附能. 全部計算工作采用Gaussian09程序完成[12], 分子的幾何構型全部由GaussView 程序從計算結構直接轉換生成, 見圖1.
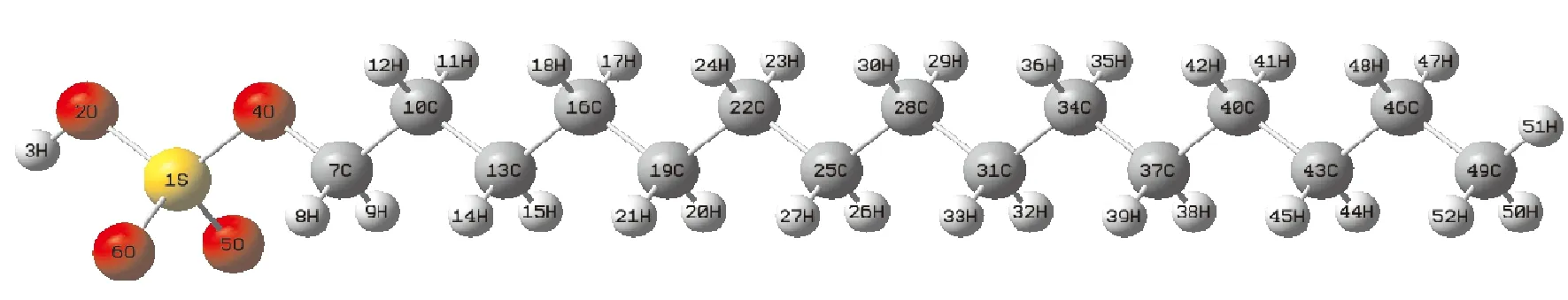
圖1 C15H32O4分子結構圖Fig. 1 The diagram of molecular structure of C15H32O4
3 結果與討論
碳原子數為15~21的硫酸氫酯類浮選藥劑離子結構和吸附銅離子后的穩定結構, 見表1.
從表1可以看出, 隨著碳原子個數增加, 硫酸氫酯類陰離子結構中碳鏈出現了彎曲. 當離子吸附銅離子后, 碳鏈也出現了彎曲, 且彎曲程度全部增大.
3.1 優化后能量變化
前線軌道理論指出, 分子的最高占據軌道(Highest Occupied Molecular Orbital, HOMO)與最低空軌道(Lowest Unoccupied Molecular Orbital, LUMO)決定著分子的電子得失與轉移, 從而決定著分子的空間取向和化學反應. HOMO上的電子能量最高, 最活潑, 也最容易失去電子, 還原性強,即HOMO 值越大, 越容易失電子, 還原性越強; 而LUMO上的電子能量最低, 最穩定, 最容易
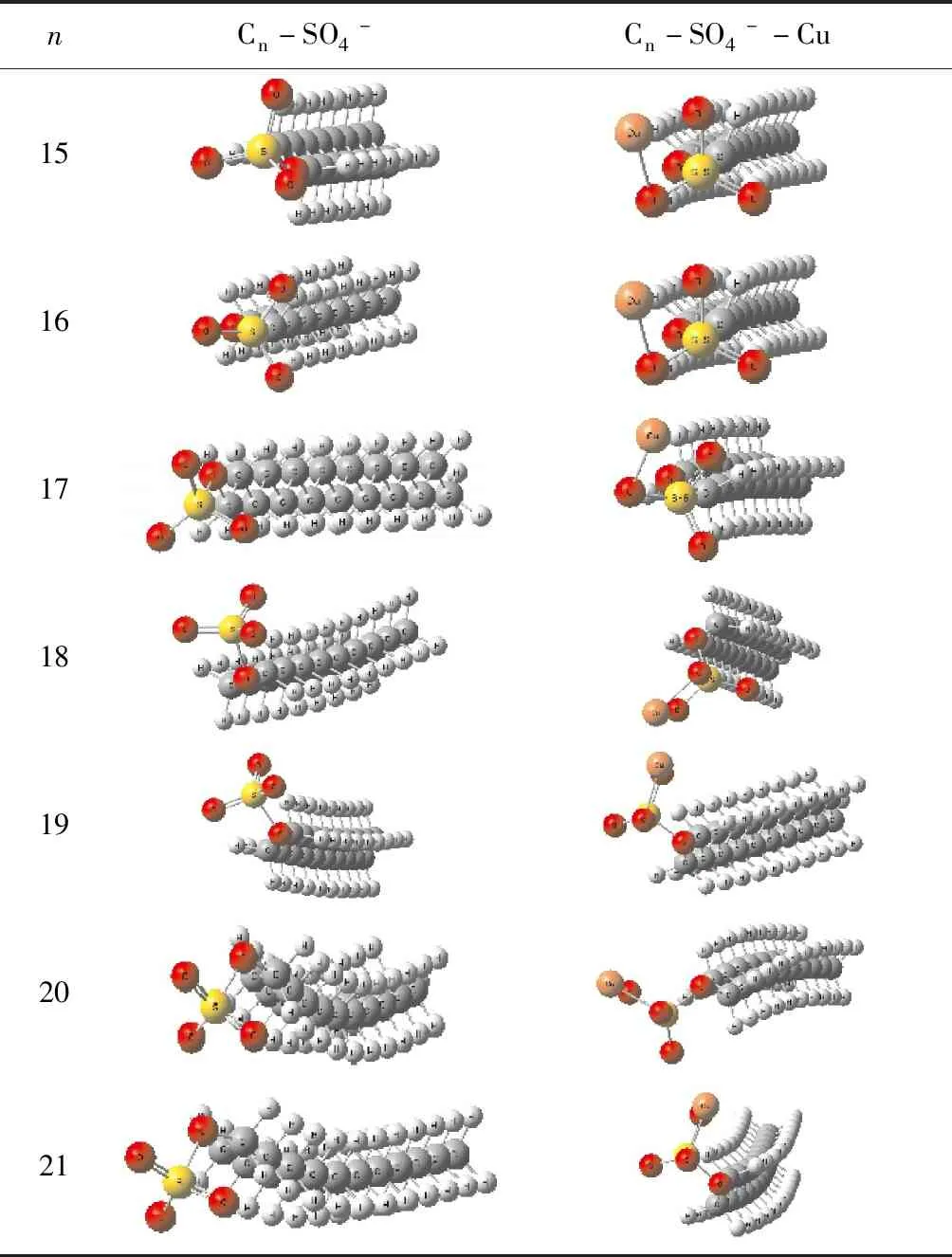
表1 分子結構圖
接受電子,具有氧化性, 即LUMO值越大, 越容易得電子, 氧化性越強[13]. DFT方法計算的碳原子數為15~21的硫酸酯類分子的前線軌道能量參數見表2, 其中n為碳原子個數,ELUMO為最低空軌道能量,EHOMO為最高占據軌道能量,ΔE1=ELUMO-EHOMO為前線軌道能量差值, 即能隙.
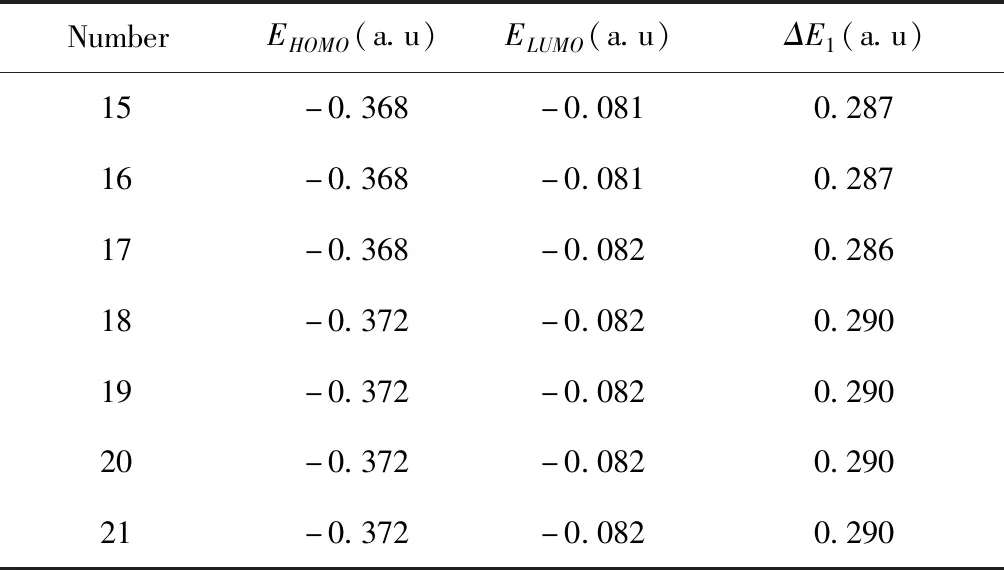
表2 硫酸氫酯的能量對比
按照化學反應性的前線分子軌道理論, 反應物的能隙是一個重要的穩定性指標,ΔE值越大, 反應物穩定性越高, 反應中活性越低, 而ΔE值小則意味著反應物易給出電子, 具有高的反應活性[3,14]. 碳原子數為15~21的硫酸氫酯類藥劑中ΔE均為正值, 該酯在吸附銅離子時都存在電子轉移, 其中C17的反應活性較強, C19~C21反應活性較低.
3.2 電荷密度分布變化
碳原子數為15~21的硫酸氫酯相同位置上的核心原子的電荷密度分布列于表3中, 吸附銅離子后, 核心原子的電荷密度分布列于表4中. 從表3可知, 在與銅離子吸附前, C15~C17硫酸氫酯相同核心原子上的電荷密度相同, 說明這三種酯對銅離子的吸附作用相同, C18~C21硫酸酯中相同核心原子上的電荷密度發生變化, 說明隨著碳原子數的增多, 酯對銅離子的吸附作用逐漸變弱[15], 與分子結構和前線軌道能量變化一致.
從表4可知, 吸附銅離子后, 電荷密度分布出現明顯變化, 相同碳原子數的硫酸酯, 硫原子和四個氧原子及和氧原子相連的碳原子上的電荷密度都出現了較大的改變. 從電荷密度分布可知, 6O上的電荷密度最小, 最容易被銅離子進攻, 即為銅離子的最佳吸附位置, 銅離子的電荷也隨著碳原子數增大而逐漸減小.
表3 硫酸氫酯核心原子的電荷分布
Table 3 The distributions of charge in the core atoms of hydrogen sulfate esters
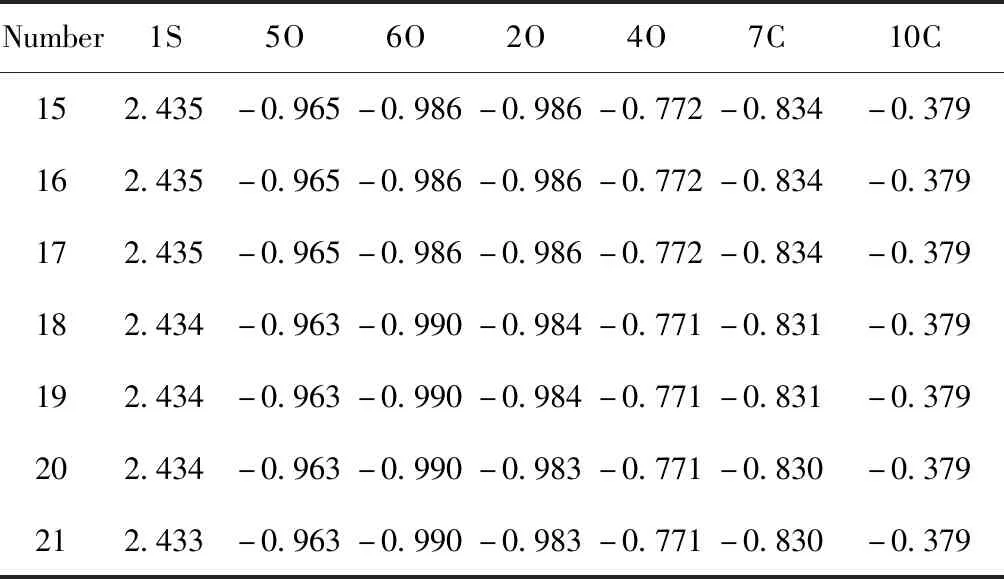
Number1S5O6O2O4O7C10C152.435-0.965-0.986-0.986-0.772-0.834-0.379162.435-0.965-0.986-0.986-0.772-0.834-0.379172.435-0.965-0.986-0.986-0.772-0.834-0.379182.434-0.963-0.990-0.984-0.771-0.831-0.379192.434-0.963-0.990-0.984-0.771-0.831-0.379202.434-0.963-0.990-0.983-0.771-0.830-0.379212.433-0.963-0.990-0.983-0.771-0.830-0.379
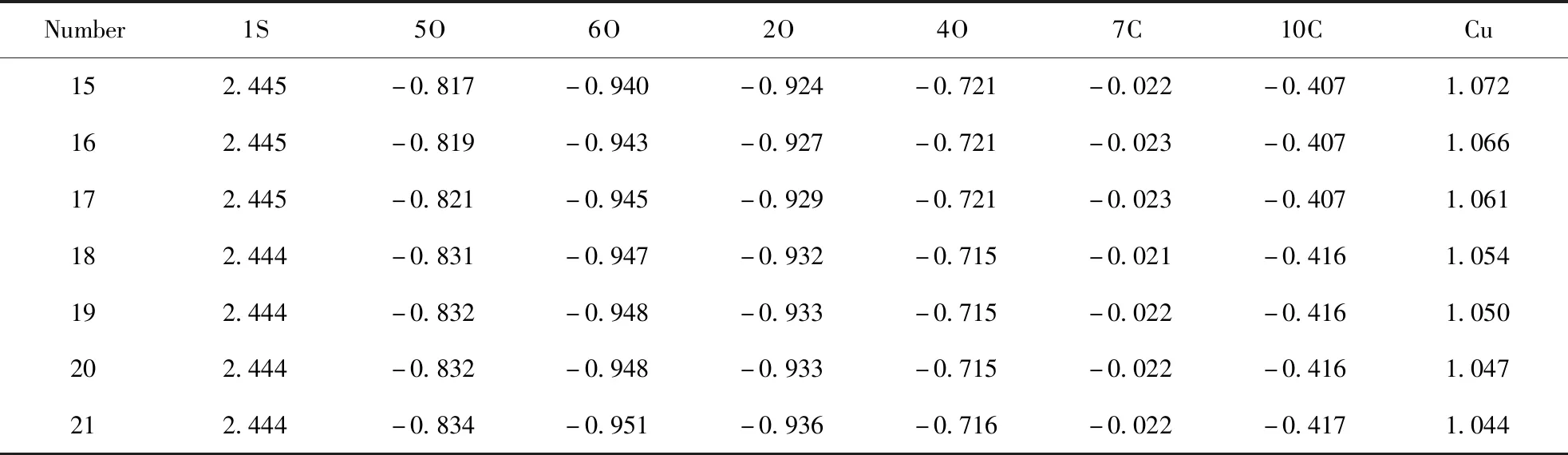
表4 硫酸氫酯吸附銅離子后核心原子的電荷分布
3.3 吸附能變化
碳原子數為15~21的硫酸氫酯與銅離子的吸附能變化列于表5中, 在ΔEabs=ECn-Cu-ECn-ECu中,ECn為不同碳原子數的硫酸氫酯的能量,ECu為銅離子的能量,ECn-Cu為不同碳原子數的硫酸氫酯與銅離子作用的吸附能.
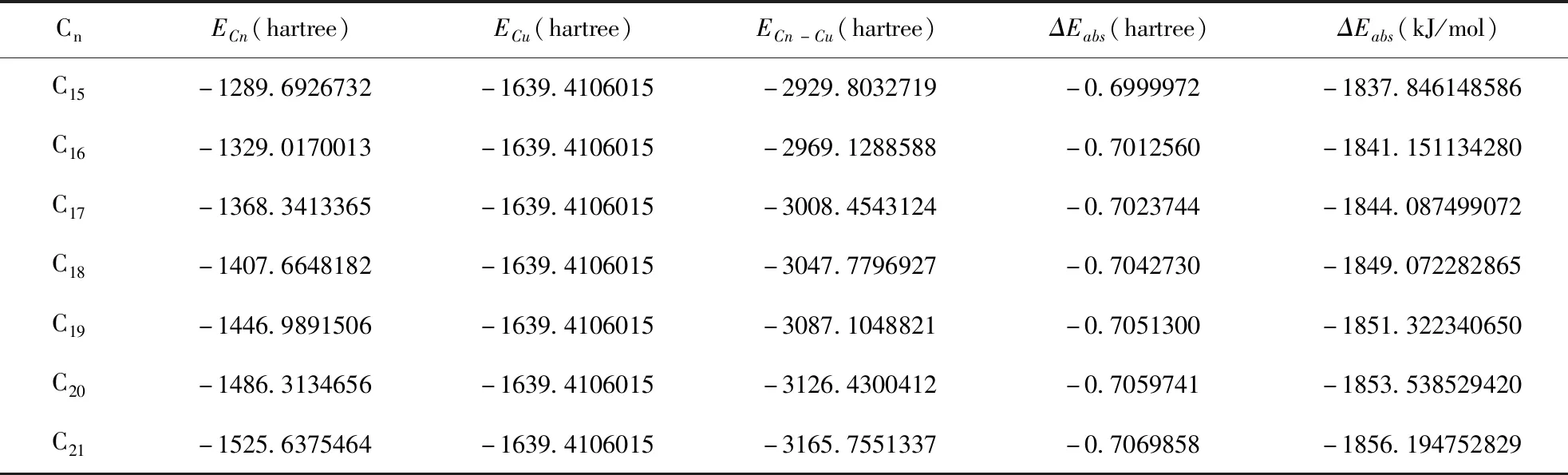
表5 硫酸氫酯與銅離子作用的吸附能
從表5可知,ΔEabs均為負, 碳原子數為15~21的硫酸氫酯均可以與銅離子發生化學吸附, 且硫酸氫酯吸附銅離子后, 總吸附能逐漸降低.
4 結 論
采用密度泛函DFT-UB3LYP方法, 6-311+G(d, p)基組, 對碳原子數在15~21的硫酸酯類浮選藥劑吸附銅離子進行了計算并得出以下結論:
(1)從前線軌道能隙來看, 碳原子數為15~21的硫酸氫酯類浮選藥劑中C17的反應活性最強, C19~C21反應活性較差;
(2)從電荷密度分布可知, 6O電荷密度最小, 是最容易被銅離子進攻的位置, 即為銅離子的最佳吸附位置;
(3)從總吸附能的計算可知, C18~C21結構較為穩定, 這與前面的電荷計算結果一致.
通過計算, 綜合考慮各種因素C17(十七烷基硫酸氫酯)浮選效果較好.
