密度泛函理論研究奈韋拉平結構,芳香性和電荷性質
2019-12-06 09:31:38郭雅晶薛乃濤李秀燕
原子與分子物理學報 2019年6期
郭雅晶,薛乃濤,李秀燕
(1. 太原師范學院物理系,晉中 030619; 2. 太原理工大學物理與光電工程學院,太原 030024)
1 引 言
奈韋拉平作為一種核苷類抗病毒藥物,特別是用作抑制藥物,在人體免疫缺陷病毒(HIV)中占有重要地位[1,2]. 一些臨床試驗證明,奈韋拉平是相當多的HIV蛋白酶的基礎抑制劑[3,4]. 一些分析表明奈韋拉平可能對這些病毒載量高或CD4計數低的患者有效[5,6]. 為此,Govindaswamy研究組從實驗角度分析了奈韋拉平上的IR、UV、量子力學和核磁共振(NMR)性質以及藥理性質[7]. Govindaswamy研究組側重于奈韋拉平的藥理性質,并且未對該物質的芳香性進行研究,基于奈韋拉平分子的特殊性,也為了進一步了解奈韋拉平的微觀物理性質和化學性質,因此從團簇水平了解奈韋拉平的結構性質是非常必要的. 所以,文中通過應用理論計算方法研究了該分子的幾何結構、芳香性和電荷性質. 據研究所知,奈韋拉平的芳香性尚未得到研究. 下文計算結果表明,該分子的所有環均具有共軛效應.
2 計算細節
所有理論計算都是在奔騰(R)/2.70GHz個人計算機上,使用Gaussian 09軟件包[8]分別在B3LYP/6-31g和HF/6-31g基組水平上運用密度泛函理論(DFT)和Hartree-Fock方法進行的. 通過使用B3LYP/6-31g和HF/6-31g基組水平上對奈韋拉平幾何結構進行了優化. 為了證實奈韋拉平團簇結構的穩定性,還分析了振動頻率. 諧波振動頻率分析也在相同的理論水平上進行,以找出勢能面上的最小值. 這里優化所得的分子具有零數量的虛頻率(NIMAG=0). 基態分子的幾何結構圖像通過GAUSSVIEW 5.0[9,10]軟件生成. 為了驗證此處使用優化水平的有效性,在理論計算中,選取了與Govindaswamy研究組[7]所得鍵長和鍵角進行了對比,發現所得的計算值與Govindaswamy研究組所得的理論計算值相比更接近Govindaswamy研究組所得的實驗值. 因此可得出,這些方法和基組水平適用于該分子體系. 隨后,采用規范不變的原子軌道(GIAO)方法研究了奈韋拉平的芳香性,在優化后的基態奈韋拉平中,鬼原子(Bq)被分別放置在四個環的幾何中心,用來計算核獨立化學位移(NICS)值. NICS是一個分子芳香性標準,它是由施萊爾提出的[11,12]. NICS負值表征芳香性,NICS正值表征反芳香性,當NICS值趨于零時,表現為非芳香性. 最后,利用自然鍵軌道(NBO)方法研究了基態奈韋拉平的電荷性質.
3 結果和討論
3.1 幾何結構
圖1給出的奈韋拉平原子幾何結構,通過HF/6-31g和B3LYP/6-31g優化所得的奈韋拉平幾何結構參數分別列于表1中. 計算得出的幾何參數(鍵長和鍵角)接近于Govindaswamy所得的實驗數據[7]. 從表1中可得出,與實驗值相比,對于奈韋拉平的鍵長,使用B3LYP/6-31g計算所得結構參數優于HF/6-31g計算所得值;對于鍵角,優化所得相關系數不如計算所得鍵長完美,例如表1中C2-C1-H7、C4-C5-N6、C17-C16-N18、C16-C17-C24、H21-C20-C24的鍵角結果. 計算所得結果與實驗值的一致性略有差異是因為程序優化是在計算機封閉隔離條件下進行的,沒有X射線和外界電壓環境的干擾.

圖1 奈韋拉平幾何結構Fig. 1 Structure of nevirapine.

表1 奈韋拉平的鍵長(?)和鍵角(°)
3.2 穩定性
表2總結了奈韋拉平的基態參數,即點對稱性、最低頻率(cm-1)、結合能(eV)、平均結合能(eV)、溫度(K)、壓力(Atm)、零點能(eV)、HOMO能級(eV)、LUMO能級(eV)和能隙(eV). 能量間隙被定義為HOMO與LUMOM差值的絕對值[8]. 在整個計算過程中,優化環境溫度為298.15 K,壓力為1 Atm. 采用DFT和HF分別計算所得的最低頻率都是正數,這意味著優化所得的幾何結構位于勢能面的局部最小值上,即該結構為基態結構. 結合能與平均結合能的均為負數,這進一步說明優化的奈韋拉平是穩定的基態結構. 表2中HOMO、LUMO和能隙的結果與Govindaswamy所得結果一致[7].
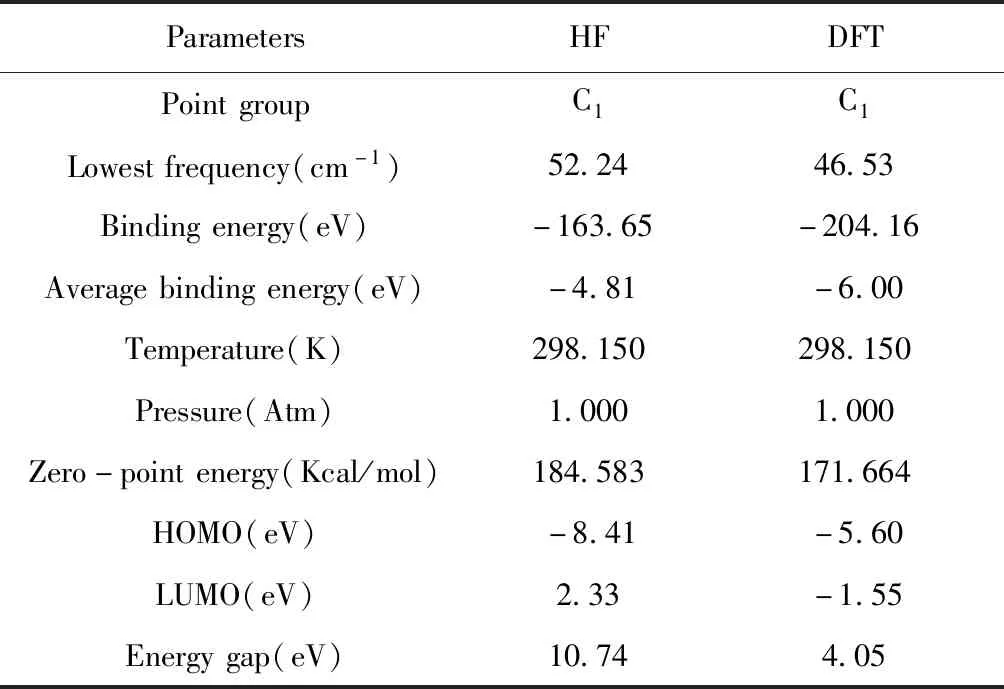
表2 奈韋拉平基態參數
3.3 芳香性
奈韋拉平的芳香性采用核獨立化學位移值(NICS)衡量,并且通過GIAO-B3LYP/6-31g和GIAO-HF/6-31g方法在基態的基礎上進行測量. 在奈韋拉平分子上設置四個參考點,每個參考位置分別放置一個鬼原子(Bq),并計算相應的NICS值. 對于表3,NICS(0.00 nm)位于奈韋拉平各環的幾何中心,NICS(0.05 nm)、NICS(0.10 nm)、NICS(0.15 nm)分別位于幾何中心垂直距離0.05 nm、0.10 nm、0.15 nm處. 結合圖1和表3,可以得出結論,環1、環3、環4具有芳香性,環2具有反芳香性,這表明四個環均為共軛體系,但是環1、環3和環4較穩定,環2能量大大提高力學穩定性較差易發生躍遷. 對于奈韋拉平,芳香性強度按環4>環3>環1的順序依次降低;另外,環1、環2和環3在NICS(0.05 nm)處芳香性和反芳香性最強.
3.4 自然鍵軌道
使用NBO方法分析基態奈韋拉平的電荷性質. 表4分別列出了使用HF和DFT理論從自然鍵軌道(NBO)計算得到的自然電子構型值和從自然總體分析(NPA)計算得到的自然原子電荷值. 由表4可看出,O15上的2s軌道所得到的電荷比2p軌道失去的電荷多,因此導致O15上呈現負電荷. N6、N13、N18、N31的2s、2p和3p軌道都從碳原子C1、C5、C10—C12、C16的2s、2p和3p軌道上獲得電荷,這導致N6、N13、N18、N31呈負電性、C1、C5、C10—C12、C16呈正電性. 此外,所有氫原子上的電荷分布基本沒有差異. 從表4和上述分析中可以得出,氮原子和碳原子形成sp2雜化,氧原子形成sp雜化.

表3 奈韋拉平基態下的核獨立化學位移值NICS(ppm)

表4 奈韋拉平的自然鍵軌道和電荷
4 結 論
分別基于密度泛函理論(DFT)和Hartree-Fock方法,結合B3LYP/6-31g和HF/6-31基組水平研究了奈韋拉平分子的基態結構性質(例如:基態結構幾何參數、結合能、能隙、芳香性、電荷性質等). 結果表明,C1點群對稱性是基態奈韋拉平的幾何結構. 對于奈韋拉平,環1、環3、環4具有芳香性,環2具有反芳香性;四個環均為共軛體系,但是環1、環3和環4較穩定,環2能量大大提高力學穩定性較差易發生躍遷;芳香性強度按環4>環3>環1的順序依次降低. 氮原子和碳原子形成sp2雜化,氧原子形成sp雜化.
猜你喜歡
房地產導刊(2022年5期)2022-06-01 06:20:14
哲學評論(2021年2期)2021-08-22 01:53:34
建材發展導向(2021年12期)2021-07-22 08:06:48
中學生數理化(高中版.高二數學)(2021年5期)2021-07-21 02:14:46
建材發展導向(2021年7期)2021-07-16 07:07:52
中學生數理化(高中版.高二數學)(2021年12期)2021-04-26 07:43:48
中等數學(2020年6期)2020-09-21 09:32:38
中華詩詞(2019年7期)2019-11-25 01:43:04
中等數學(2019年6期)2019-08-30 03:41:46
中學生數理化·七年級數學人教版(2018年4期)2018-06-28 03:26:30
