堿水解-SPE-LC-MS/MS法快速測定動物源食品中喹乙醇代謝物殘留量
2019-12-19 07:56:18余婷婷
食品與機械 2019年11期
關鍵詞:檢測
劉 迪 韓 莉 曾 妮 余婷婷 -王 亨 王 彬 榮 茂
(1. 湖北省食品質量安全監督檢驗研究院,湖北 武漢 430075;2. 湖北省食品質量安全檢測工程技術研究中心,湖北 武漢 430075)
喹喔啉類藥物是一類包含有喹喔啉-1,4-二氮氧結構[1]通過化學合成的專用獸用藥物,因其具有良好的抗菌和促進生長的效果,被廣泛添加于禽、畜和水產養殖飼料中[2],其中喹乙醇是喹喔啉類藥物的典型代表[3]。動物基體對于喹乙醇有蓄積作用,并且會引起動物產生畸形,對人類更是有致突變和致癌的作用[4]。喹乙醇原藥的性質不穩定,在體內代謝迅速,能產生多種代謝產物,其中的絕大多數性質也不穩定,無法被定量檢測。3-甲基-喹喔啉-2-羧酸(MQCA)作為喹乙醇的主要代謝產物之一,因其代謝過程比較長,并且殘留量相對穩定,現在通常都是通過測定MQCA的含量來間接反映喹乙醇的殘留情況[5]。中國自2019年5月1日起停止經營、使用喹乙醇獸藥的原料藥及各種制劑[6],而早在1998年,喹乙醇就已經被歐盟禁止作為飼料添加劑[7]。時至今日,MQCA用很多檢測方法可以檢測[8-12],其中液相色譜法和液相—質譜/質譜為主流的檢測方式。動物源食品基質復雜,含有大量的脂肪、蛋白質等物質,液相色譜法一般需要經過相對復雜的前處理,且定性難度較大,容易產生假陽性;而液相—質譜/質譜檢出限低,抗干擾能力和定性能力較強,可以在降低檢出限的同時避免假陽性的產生。MQCA的前處理方式通常為酶解和水解(酸解),酶解耗時長,酸解提取效率不高,后續凈化效果不徹底,檢出限高,靈敏度不足。采用液相—質譜/質譜檢測也需要不斷地改進優化前處理方法,縮短樣品測定時間,減少基質效應,提高靈敏度的同時保證準確度,李佩佩等[13]用免疫親和柱對MQCA進行特異性吸附,效果較好,但免疫親和柱價格成本較高,且容易堵塞[14]。痕量污染物分析檢測的重點是簡化操作,減少干擾,以期提高靈敏度;而難點是分離分析物和雜質以及合適的凈化方法[15]。張小軍等[16]采用鹽酸水解提取MQCA,陰離子固相萃取柱凈化,對魚肉等水產食品的水解程度可以達到很好的效果,但對于畜禽肉水解效果并不是很理想;吳玉杰等[17]對水解液進行反復的液液萃取,操作繁復,過程損失大。
試驗擬建立堿水解提取,固相萃取法凈化和液相串聯質譜聯用的3-甲基-喹喔啉-2-羧酸的分析方法,為快速檢測和監管動物源性食品中3-甲基-喹喔啉-2-羧酸殘留提供技術保障和參考。
1 試驗部分
1.1 材料及試劑
雞肉、豬肉、魚:武漢市售;
陰性基質:通過國家標準方法測定驗證;
3-甲基-喹噁啉-2-羧酸標準品:純度均≥99%,德國Dr.Ehrenstorfer公司;
喹噁啉-2-羧酸-D4(quinoxaline-2-carboxylic-D4,QCA-D4)標準品:純度≥99%,德國Witega公司;
正己烷、鹽酸、氫氧化鈉:分析純,上海國藥集團化學試劑有限公司;
乙腈、甲酸、甲醇、乙酸乙酯:質譜級,德國默克公司;
Oasis MAX固相萃取柱:美國Waters公司;
試驗用水:去離子水(電阻率≥18.2 MΩ·cm),Millipore-Q超純水儀自制。
1.2 儀器與設備
串聯四級桿質譜儀:Xevo TQD型,美國Water公司;
液相色譜儀:ACQUITY UPLC型,美國Water公司;
高速離心機:Allegra X-15R型,美國Beckman Coulter有限公司;
電子分析天平:XS204、ME2002E型,梅特勒—托利多儀器(上海)有限公司;
旋轉蒸發器:HEI-VAP/LR20型,德國Heidolph GmbH公司;
超聲波清洗器:Elmasonic P型,德國艾爾瑪公司;
漩渦混合儀:EOFO基礎型,美國Talboys公司;
水浴氮吹儀:N-EVAP24型,美國Organomation公司。
1.3 標準溶液的配制
在兩個10 mL容量瓶中分別準確稱量10 mg MQCA和QCA-D4標準品,加入甲醇溶解并稀釋至刻度,配制成質量濃度為1 mg/mL的MQCA標準儲備液和1 mg/mL QCA-D4同位素內標儲備液。放置于-20 ℃的冰箱,有效期3個月。根據需要,用甲醇將MQCA和QCA-D4標準儲備液稀釋成濃度均為1 μg/mL中間標準溶液。臨用前,將中間標準溶液用0.1%甲酸水溶液配制成0.5~50.0 ng/mL 的同位素內標曲線,每毫升該標準工作液含有同位素內標5 ng。
1.4 樣品處理
稱取5.00 g搗碎并已均勻混合的樣品到50 mL的聚丙烯離心管中,然后加入50 μL QCA-D4同位素內標標準溶液,加入2 mol/mL NaOH溶液20 mL,渦旋均勻,60 ℃ 水浴條件下水解1 h[18]。待水解液冷卻至室溫后,用10 mol/L HCl溶液將水解液pH值調至1.0±0.2,4 000 r/min 離心5 min,上清液轉移至50 mL離心管中,加入20 mL正己烷,充分混合,4 000 r/min離心5 min,去除正己烷層。向離心管中加入乙酸乙酯15 mL,渦旋混勻器上渦旋3 min,4 000 r/min、4 ℃條件下離心5 min,將上層乙酸乙酯轉移至50 mL雞心瓶中。提取液用15 mL 乙酸乙酯復提一次,合并乙酸乙酯于同一雞心瓶中,50 ℃下旋轉蒸發至凈干,用5 mL 2%氨水溶液溶解殘渣,待凈化。用3 mL甲醇和3 mL水活化Oasis MAX固相萃取柱后,所有待凈化的液體均經過MAX固相萃取柱,等待樣品溶液完全流出后,用3 mL 0.05 mol/L氫氧化鈉溶液,水和甲醇溶液依次淋洗, 將流出液全部棄去。 加壓吹干2 min,并用3 mL 2%的甲酸甲醇溶液洗脫,洗脫液收集于15 mL離心管中[19]。洗脫液置于50 ℃水浴下氮氣吹干,將殘余物用1.0 mL的0.1%甲酸水復溶,0.22 μm 有機濾膜過濾,供液相串聯質譜檢測。內標法定量。
1.5 液相色譜條件
色譜柱型號:Waters ACQUITY UPLC BEH C18色譜柱(2.1 mm×50 mm,1.7 μm);色譜柱溫度:35 ℃;進樣量:5 μL;流速:0.30 mL/min;流動相A:0.1%甲酸水溶液,流動相B:乙腈;梯度洗脫過程:0.0~1.0 min,98% A;1.0~2.5 min,98%~50% A;2.5~2.6 min,50% A;2.6~3.0 min,50%~10% A;3.0~3.2 min,10%~98% A;3.2~5.0 min,98% A。
1.6 質譜參考條件
離子模式:正離子模式;離子源類型:電噴霧離子源(ESI源);離子源溫度:120 ℃;掃描方式:多反應監測(MRM)模式;毛細管電壓:2.00 kV;去溶劑氣:高純N2,流速:800 L/h,溫度:450 ℃;錐孔氣:高純N2,流速50 L/h;碰撞氣:高純Ar。
2 結果與分析
2.1 質譜條件的確定
分別在ESI+和ESI-模式下,根據目標化合物的性質和結構,選擇響應最高的離子加和峰作為母離子,通過對比ESI+和ESI-模式下的母離子響應,確定試驗在ESI+下進行。對選定的母離子加碰撞能量擊碎,同時進行離子掃描,分析可能的斷裂規律以及根據二級碎片離子掃描質譜圖,分別選取離子豐度相對較強的一對碎片離子作為定量離子,次強的一對碎片離子作為定性離子,最后在多反應監測模式(MRM)下分別對錐孔電壓、碰撞能量進行優化。對于同位素內標物質而言,僅需一個豐度相對最強的一對碎片離子即可。表1中顯示了最終確定多反應監測條件的質譜參數。
表1 3-甲基-喹噁啉-2-羧酸和內標物的離子對及錐孔電壓、碰撞能量?
Table 1 Ion pair and cone voltage and collision energy of MQCA and internal standard
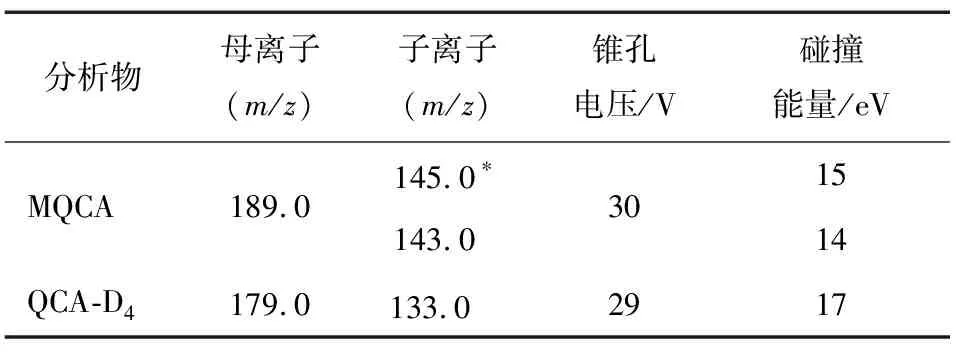
分析物母離子(m/z)子離子(m/z)錐孔電壓/V碰撞能量/eVMQCA189.0145.0?143.0301514QCA-D4179.0133.0?2917
? 帶*的離子為定量離子。
2.2 色譜條件的確定
MQCA為極性一般的弱酸性化合物,在C18柱上有好的保留效果[8],因此采用Waters ACQUITY UPLC BEH C18(2.1 mm×50 mm,1.7 μm)的超高效液相色譜柱進行試驗。同時,在正離子模式下以乙酸銨溶液作為流動相,會出現一定的離子抑制作用;而流動相中添加一定量的甲酸,減小pH值的同時,不僅可以改善峰型,還可以提高離子電離效率以及目標與雜質的分離度。通過對0.1%甲酸溶液與乙腈、甲醇分別作為有機相組成流動相的效果進行了比較。對于標準品而言,乙腈和甲醇均有較好的峰型,甲醇作為流動相時的峰寬要大于乙腈作為流動相時的峰寬;并且分離樣品時,以乙腈作為流動相時不僅靈敏度略高,并且目標物和雜質的分離效果明顯要比甲醇作為流動相時的目標物和雜質的分離效果好,因此試驗選擇流動相為乙腈—0.1%甲酸溶液。色譜條件經過優化后,MQCA的峰形得到改善,與基質中的雜質也有較好的分離度,峰型尖銳,整個分離過程可以在5 min內完成。標準溶液、空白基質樣本及添加藥物樣本的選擇離子流色譜圖見圖1~3。
2.3 水解方式的選擇
水解方式一般為酶水解[20]、酸水解[21]、堿水解[22]3種,MQCA與其他獸殘檢測一樣,也是先水解組織(破壞目標物與細胞的結合),再凈化提取液,然后再檢測凈化液的流程。酶水解過程十分溫和,需在(47±3) ℃振蕩水浴中反應16 h以上,酶解過程耗時長且步驟繁瑣;酸水解反應相對較溫和,所以對于豬肉等樣品,其水解效率不高。因為MQCA與組織主要是共價結合,采用酶解和酸解的方式并不能完全將MQCA從組織中解離出來[18]。強堿水解相對于酶解和酸解效果更好,反應通常較為劇烈,能在短時間內有效水解樣品中的蛋白質并將MQCA從組織中解離出來。試驗通過改變堿水解時間以及NaOH堿溶液的濃度,通過水解液狀況評價水解效果。選擇水解時間分別為20,40,60,80,100,120 min,NaOH溶液濃度分別采用0.2,0.5,1.0,2.0,5.0,10.0 mol/L,在60 ℃下對樣品進行水解。通過對比發現,NaOH濃度太低,水解過程耗時長;NaOH濃度越高水解液相反會變得濃稠,不適合后續的提取凈化過程。選用2 mol/L NaOH溶液水解60 min時,基質樣品被完全水解,MQCA從樣品中被完全釋放出來,沒有殘渣剩余。因此,選擇該條件進行后續試驗。
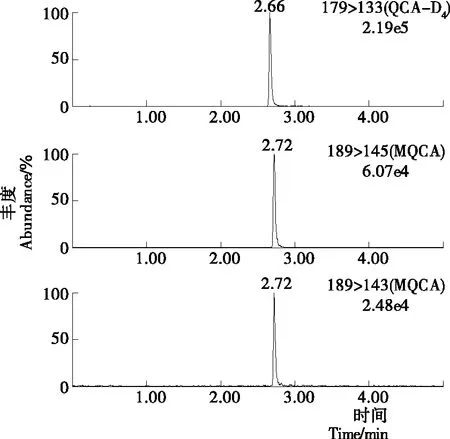
圖1 標準溶液選擇離子流色譜圖

圖2 空白基質樣本選擇離子流色譜圖

圖3 添加藥物樣本的選擇離子流色譜圖
2.4 凈化條件的確定
動物源樣品通常包含大量的蛋白質、脂肪等內源性物質,直接檢測必定會產生基質效應,對檢測造成影響,對設備也會造成損害。常用凈化的方式有固相萃取法和液液萃取法,試驗采用液液萃取法與固相萃取法結合凈化的方式。大多數的文獻和國家標準都是選擇的MAX固相萃取小柱[19,21-23],因為MQCA能解離形成負離子,可以與MAX上的季銨正離子發生相互作用結合。試驗發現,水解液pH較大,濾液比較黏稠,若不經過處理直接過固相萃取柱容易堵塞固相萃取小柱篩板,使過柱流速變慢,導致后續凈化過程無法進行,因此必須先通過液液萃取過程,將目標物萃取出來。由于MQCA呈弱酸性,水解液必須要經過酸化后MQCA才能形成分子形態,才能被乙酸乙酯萃取,并且MQCA分子在堿性條件下才能夠離解形成負離子,與MAX上的季銨正離子才能充分鍵合。試驗選擇用濃鹽酸對水解液進行酸化,強酸對蛋白起到一定的沉淀作用,乙酸乙酯萃取MQCA,濃縮乙酸乙酯后,用2%氨水溶液溶解殘渣,通過MAX固相萃取小柱凈化,整個過程符合陰離子固相萃取柱的陰離子交換作用的原理,可以達到理想的富集和凈化的效果。
2.5 基質效應
將MQCA標準溶液分別用流動相和空白基質提取溶液稀釋成0.5,1.0,2.0,5.0,10.0 ng/mL,均以峰面積為縱坐標,MQCA質量濃度為橫坐標作圖,即可分別得到標準工作曲線和基質曲線,通過兩個曲線的斜率的比值可知,MQCA的基質曲線響應值約為標準曲線響應值的60%,說明MQCA在所驗證基質中有一定程度的離子抑制效應,檢測過程中的基質抑制效應和試驗提取過程中的損耗會對定量帶來誤差。為了定量更加精準,在實際樣品檢測中既可采取基質匹配外標曲線校正法,也能采用同位素內標標準曲線法進行定量。鑒于基質匹配外表曲線校正法的操作相對比較麻煩,試驗采用同位素內標標準曲線法作為最終的定量方法,MQCA和QCA化學性質相近,參考相關標準和文獻[19,23-24],選擇QCA-D4作為內標。
2.6 線性關系和檢出限
根據上述所確定的液相條件、質譜參數及構建的MQCA殘留的檢測分析方法,對配制的0.5~50.0 ng/mL的MQCA系列標準工作曲線進行方法學指標分析。結果表明,在0.5~50.0 ng/mL濃度范圍內,MQCA呈良好的線性關系,擬合曲線為y=0.659 6x+0.085 5(R2=0.999 7)。取陰性樣品,通過加入低濃度MQCA標準溶液制得濃度含量低的加標樣品,按照1.4方法對樣品進行處理,以3倍信噪比計算方法的檢出限,根據結果,確定此方法MQCA檢出限為0.1 μg/kg (S/N=3)。
2.7 回收率和精密度
分別準確稱取5.00 g豬肉、雞肉、魚肉搗碎樣品(陰性),分別加入MQCA和QCA-D4標準溶液,得到0.1,0.2,1.0 μg/kg 3個濃度添加水平的加標樣,每個濃度水平做6個平行。所得樣品按照1.4方法處理,通過測定濃度與添加濃度之比計算回收率,并通過6次測定的回收率得出相對標準偏差。各基質加標回收率和相對標準偏差見表2。由表2可知,動物源食品中目標化合物的回收率為95.6%~108.2%,相對標準偏差為3.4%~14.3%(n=6)。
表2魚,雞肉和豬肉中MQCA的加標回收率和相對標準偏差
Table 2 Additive recovery and relative standard deviation of MQCA in fish,chicken and pork(n=6)
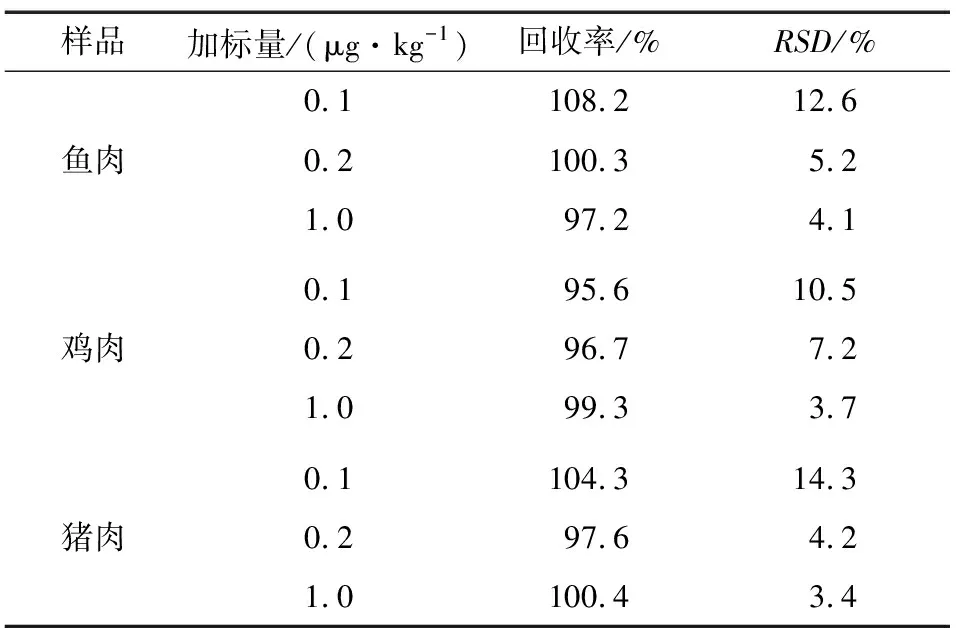
樣品加標量/(μg·kg-1)回收率/%RSD/%0.1108.212.6魚肉0.2100.35.21.097.24.10.195.610.5雞肉0.296.77.21.099.33.70.1104.314.3豬肉0.297.64.21.0100.43.4
2.8 市售動物源食品的測定
采用試驗方法對流通市場中的30批動物源食品樣品(魚、雞肉、豬肉樣品各10個)進行檢測,結果表明,魚肉和雞肉樣品中均未檢出MQCA;有2個批次的豬肉中檢出了MQCA,其殘留量分別為0.23,3.08 μg/kg(根據GB/T 20746—2006國家標準方法測定的結果則為未檢出和2.87 μg/kg)。綜上可知,喹乙醇藥物在中國仍在使用并存在組織殘留的風險,在一定程度上對食品安全造成了威脅。漁業養殖和禽類養殖過程中對于喹喔啉類藥物的使用和監管有了一定的成效;而由于喹乙醇代謝物在豬肉中存在允許限量,豬飼料里面允許加入適量的喹乙醇,所以豬肉中相對檢出率要高。
3 結論
通過在堿性環境下水解提取樣品組織,MQCA能夠有效地從組織中游離,通過調節pH值在酸性介質下液液萃取提取MQCA,經過混合型強陰離子交換小柱凈化等樣品前處理技術除去大量雜質,減少基質效應,建立了一種快速、準確檢測喹乙醇代謝物MQCA在動物源性食品中殘留量的LC-MS/MS方法。該方法分離效果良好,操作簡單快捷,準確度高,精密度較好。在0.1~1.0 μg/kg的添加水平范圍內的平均回收率為95.6%~108.2%;相對標準偏差為3.4%~14.3%(n=6)。該方法適合動物源食品中MQCA殘留的快速定量定性分析。運用高靈敏度和強特異性的免疫學方法的對大批量樣品進行盲篩,以及結合液相串聯質譜的強定性定量能力,發展基于新型材料更便捷的前處理方法,是今后亟需探索和發展的新方向。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
