細胞衰老的機制概述
2019-12-27 06:21:14馮春艷陳旭遠周海城
生物學教學 2019年12期
馮春艷 陳旭遠 林 闖 周海城
(1 東北師范大學教育學部 長春 130024; 2 吉林省實驗中學 長春 130022)
衰老的細胞通常具備核膜內折、染色質收縮、脂褐質增多、膜結構改變、物質運輸功能降低、細胞器結構和功能異常等特征,而這些相對于健康的細胞而言都是損傷,也就是說細胞衰老是損傷通過時間的累積而使之熵變再經過自然選擇的結果。目前,對細胞衰老原因的討論主要集中在氧化應激產生自由基、端粒縮短、DNA損傷反應(DDR)、胰島素/IGF-1代謝異常、細胞自噬異常這幾個方面。從各種衰老機制的發生序列和損傷累積的視角來看,自由基和端粒縮短對細胞的損傷是衰老的伊始,DDR和胰島素/IGF-1代謝途徑是稀釋損傷的信號轉導通路,細胞自噬則是清除損傷的過程。
1 細胞損傷: 衰老伊始
當外界壓力因素,如輻射、紫外線、重金屬作用于細胞時,細胞中的DNA、蛋白質、膜結構或細胞器可能會受到損傷。這些因應激反應產生的變化不斷增加,成為“損傷累積”,進而導致有機體無序性的增加,即熵增。任何生物都存在著產生有缺陷產品的風險,細胞代謝過程中產生自由基等有毒的副產物,如氧在正常的代謝過程中產生包括過氧化物、超氧化物等活性氧(ROS),應激條件下ROS會增多,進而損害脂質、核酸、蛋白質和細胞器,觸發相應修復反應[1]。
端粒是存在于真核生物染色體兩端高度保守的非編碼重復DNA序列。端粒DNA與Shelterin蛋白(包括TRF1、 TRF2、 Rap1、 TIN2、 TPP1、 POT1)一起存在于真核染色體的末端形成“帽”,從某種意義上來看,端粒是在用重復序列封閉每條染色體,保護染色體免受縮短造成的損害[2],同時也能夠避免染色體之間錯亂的融合。研究發現: 隨著細胞復制次數的增加,端粒DNA就會縮短一截,使細胞復制達到海弗利克極限(Hayflick limitation),最終導致細胞衰老和死亡。端粒長度與潛在的壽命變化有關,端粒的縮短會使染色體對輻射的敏感度增加,從而增加了變異的可能性[3]。端粒酶可以阻止端粒丟失,但端粒酶在大多數體細胞組織中都處于失活狀態。從生命進化的視角來看,端粒的縮短、細胞的衰老都是最終所選擇下來的生存機制,細胞衰老雖然限制了細胞分裂的次數,但避免了細胞無限分裂成為癌細胞。
2 稀釋損傷: 信號轉導
當細胞受到損傷之時,細胞內便會啟動DNA的受損修復機制、蛋白質等分子結構的修飾以稀釋損傷。損傷稀釋可以抵消有害變化的累積,特別是當細胞分裂非常迅速并且它們獲得的損傷相對溫和時[4]。當損傷變得難以稀釋時,損傷就會更容易附著到某個細胞結構上,從而隨著細胞分裂而不對稱的分散它們[5]。通常來說,在轉錄水平通過高度保守的信號轉導途徑是協調許多基因激活以延長壽命的關鍵[6]。當前研究中對細胞內損傷稀釋的機制主要包括DDR和胰島素/IGF-1信號轉導通路。
2.1 DDR信號轉導通路 DDR是一個由眾多蛋白質構成的網絡信號轉導路徑,能夠感應由不同刺激引發的細胞損傷,阻滯細胞周期的信號轉導通路。當DNA損傷發生時就會啟動DDR,引發激酶級聯增強反應,通過效應器p53、細胞分裂周期蛋白25(cell division cycle 25, CDC25)、染色體結構維持蛋白1(structural maintenance of chromosomes proteins 1, SMC1)等使細胞周期停滯,DNA損傷修復通路活化,細胞衰老或死亡[7]。
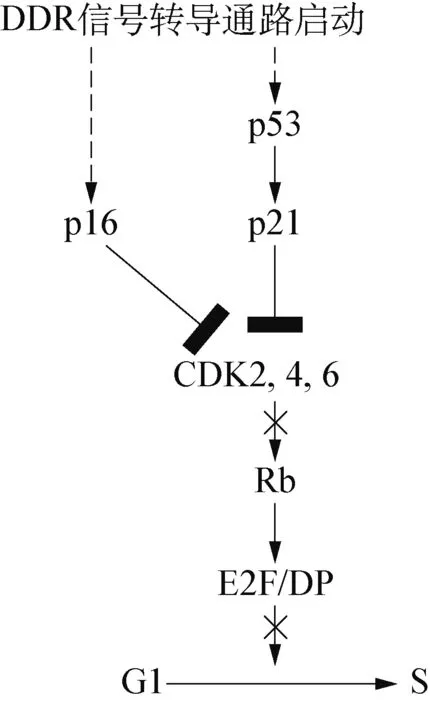
圖1 DDR信號轉導通路
細胞內的p21是由Cdknla編碼的細胞周期調控產物,是轉錄因子p53基因的下游產物。當DDR發生時,誘導p53的表達,p53通過識別失去功能的端粒,繼而誘導p21的表達,p21使得若干cyclin依賴性的激酶(CDK),如CDK2、 CDK4、 CDK6等失活,從而阻止Rb蛋白的磷酸化,Rb蛋白不能與E2F分離,E2F處于持續失活狀態,不能起始G1/S轉換過程中若干關鍵因子的轉錄,導致細胞周期停滯在G1/S期,最終引發細胞衰老(圖1)[8]。除了p21抑制CDK磷酸化的路徑外,在一些研究中表明p16也能抑制CDK4和CDK6的活性,并且DDR能夠獨立激活p16基因的表達[9]。當DNA受損或DNA復制造成端粒縮短時,就會被機體識別為DNA雙鏈斷裂(DNA double-strand breaks, DSBs)損傷,啟動DDR,阻滯細胞周期在G1/S期,導致細胞衰老或死亡,還可能在表現型上發生改變,如個體衰老、疾病產生等。
2.2 胰島素/IGF-1途徑 近些年的研究發現,影響動物衰老的一個重要的代謝途徑是胰島素/IGF-1。IGF-1又被稱為胰島素樣生長因子1,由肝臟產生,調節DNA的合成、刺激全身細胞生長[10]。胰島素/IGF-1信號(IIS)是生長和代謝的核心,在秀麗隱桿線蟲中,IIS的減少增加了抗逆性和壽命。有研究顯示胰島素/IGF-1途徑感知營養素、飲食限制和降低IIS被認為是促進蛋白水解、延緩細胞損傷累積的重要途徑。改變胰島素/IGF-1代謝途徑可導致細胞或生物體代謝狀態的全局變化,在已有大量損傷的前提下,通過減慢代謝來改變總體損傷累積速率。研究表明通過胰島素/IGF-1樣受體途徑發出信號是許多生物衰老的重要原因[11]。長壽命的IIS突變體在整個生命過程中顯示出非常低的蛋白質合成和降解水平,這種突變體壽命的延長強烈依賴于自噬途徑。
3 清除損傷: 細胞自噬
自噬是細胞維持內部穩態的一種方式,也是保證細胞長壽的重要機制之一,細胞內損傷的蛋白質、細胞器、脂質如果不能被自噬所清除,就無法為細胞代謝活動和新原料的存儲提供場所。自噬分為巨自噬(macroautophagy, MA)、微自噬(microautophagy)、伴侶蛋白介導自噬(chaperone-mediated autophagy, CMA)三種,巨自噬指的是損傷的細胞器形成自噬體最后被溶酶體降解,微自噬指的是溶酶體直接吸收細胞溶質并將其分解,分子伴侶介導的自噬(CMA)則是特定蛋白被靶向運輸到溶酶體[12]。在CMA中,熱休克蛋HSC70、Hsp90和小的熱休克蛋白家族作為分子伴侶參與自噬過程,HSC70識別帶有KFERQ序列的可溶性胞質蛋白底物,引導底物進入溶酶體。如果HSC70受損,其執行的自噬功能就會降低,細胞衰老的風險就會增加。同樣,如果衰老的線粒體不能順利被自噬,將會使細胞處于高濃度的有毒物質(包括ROS)的風險之中,影響細胞的代謝和壽命。研究發現有缺陷的線粒體很少自噬,從而ATP產生減少,細胞分解代謝機制崩潰,最終細胞衰老和死亡[13]。一般細胞自噬功能降低會引起各器官功能衰退、機體老化及相關疾病發生,增強細胞自噬功能可延緩衰老。所以,衰老細胞器的自噬是減弱氧化應激反應對細胞結構造成傷害的過程,也是細胞內穩態得以維持的重要機制。
當細胞受到損傷之初,如果衰老細胞通過細胞凋亡去除,則很難看出衰老細胞如何導致長期損傷。如果生物體不能進行及時地迭代更新或稀釋損傷的話,細胞內損傷就會累積。一旦細胞內的損傷繼續累積,就必然會對細胞產生有害的影響,并可能累及其他細胞;如若無法通過細胞自噬及時清除損傷,那么最終將會導致生物體衰老或者疾病。
猜你喜歡
人人健康(2023年26期)2023-12-07 03:55:46
中學生數理化·七年級數學人教版(2019年10期)2019-11-25 07:33:58
中國生殖健康(2019年2期)2019-08-23 08:12:10
中學生數理化·高一版(2018年9期)2018-10-09 06:46:50
湖南教育·C版(2018年3期)2018-06-05 16:54:36
中國衛生(2016年3期)2016-11-12 13:23:26
中國衛生標準管理(2015年1期)2016-01-14 03:41:27
藥學與臨床研究(2015年4期)2015-06-05 11:35:51
中國醫藥科學(2015年15期)2015-02-27 12:32:27
中國衛生(2014年12期)2014-11-12 13:12:52
