ICP-MS測定優級純無機酸中38種痕量金屬元素
2020-03-09 06:55:08袁永海李政林余紅霞丁姍姍
桂林理工大學學報 2020年4期
楊 鋒,袁永海,李政林,余紅霞,丁姍姍
(桂林理工大學 廣西隱伏金屬礦產勘查重點實驗室,廣西 桂林 541006)
0 引 言
地質樣品的化學成分分析包括主量和次量組分分析。主量組分的元素分析時,由于分析對象的含量較高,分析純的試劑即可滿足測試要求;但在進行次量組分中微量和痕量元素測試時,待測元素的含量一般為10-9或10-12級,試劑中的雜質元素往往會對測試結果帶來顯著的正干擾。特別是在進行高精度金屬同位素組成分析時,測試過程中使用的儀器(如多接收器質譜儀)對所引入樣品的純度有極高的要求,否則將會造成同位素數據的準確度和精密度達不到要求。地球化學中金屬同位素分析一般要求被測元素的全流程本底低于測試總量的0.5%[1],而流程本底的高低取決于實驗室環境、器皿和試劑;在這3個因素中,試劑本底的影響最大。試劑主要來自樣品前處理過程中用到的鹽酸、硝酸和氫氟酸,這3種優級純的酸中金屬雜質含量通常低于1 μg·L-1,很難準確測定其結果。電感耦合等離子體質譜技術(ICP-MS)可以滿足這一測試需求, 它將電感耦合等離子體的高溫電離特性(約8 000 K)與高靈敏質譜計的快速掃描相結合, 是當前最強有力的微量及痕量金屬元素分析技術, 具有多元素同時分析、 檢出限低、 干擾少、 精密度高和線性范圍寬等優點, 已被廣泛應用在地質、 環境、 冶金、 核工業、 生物、 醫藥等領域[2-9]。然而,即使ICP-MS的靈敏度高,直接將無機酸樣品稀釋至2%左右進行上機測試其中的雜質元素含量也往往達不到儀器的檢出限。此外,由于ICP-MS的截取錐和采樣錐一般為鎳或鉑金制品,濃度過高會嚴重影響錐的使用壽命。
相關的文獻報道大多是基于膜去溶進樣系統或高分辨質譜技術。如陳黎明[10]利用膜去溶進樣系統直接進樣,高分辨ICP-MS測定了半導體級高純硝酸中的25種痕量金屬雜質,方法的檢出限為0.69~23.73 ng·L-1。李春華等[11]同樣采用高分辨質譜對電子級氫氟酸中的關鍵雜質元素砷、磷、硼、鋅進行了測定,針對不同元素采用了不同的分辨率模式來消除多原子干擾, 各元素的檢出限均小于5.00 ng·L-1。而膜去溶進樣系統和高分辨質譜費用和維護成本較高,普通實驗室不具備這樣的條件。基于此,本文提出了一種利用普通的ICP-MS測定無機酸中雜質元素的方法,即通過對無機酸樣品的預富集,然后轉成硝酸介質(以降低氯化物多原子的干擾和氟離子對儀器石英管的損壞);在碰撞反應池模式下,利用ICP-MS對3種優級純無機酸中的38種痕量金屬元素進行準確測定,并對二次純化和優級純氫氟酸中的雜質元素含量進行了對比。
1 實驗部分
1.1 主要儀器與試劑
實驗采用配有碰撞反應池的電感耦合等離子體質譜儀(美國Agilent公司,型號7500cx),儀器工作參數見表1。
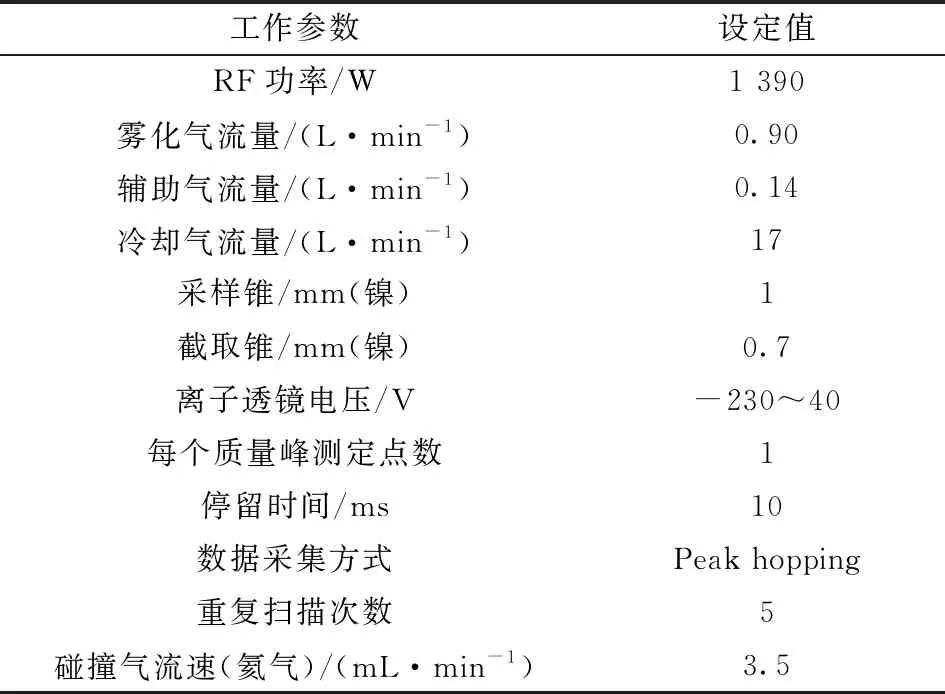
表1 ICP-MS(Agilent-7500cx)儀器工作參數
使用的優級純鹽酸、氫氟酸、硝酸為廣州市西隴化工有限公司生產。高純硝酸是優級純硝酸經過美國Savillex DST-1000 PFA亞沸蒸酸裝置在亞沸狀態下二次純化制備。實驗用水為電阻率為18.2 MΩ·cm超純水(由美國Millipore公司的純化系統純化)。
所用30 mL帶蓋Telfon器皿均經過50%鹽酸、50%硝酸和超純水反復煮沸清洗。電熱板為濱海縣正紅塑料儀器廠生產的DBF系列分體式防腐電熱板。
所用的GSB 04-1767—2004、GSB 04-1768—2004、GSB 04-1789—2004標準溶液為國家有色金屬及電子材料分析測試中心提供的混合標準溶液, Mg、 Ti、 V、 Cr、 Mn、 Fe、 Co、 Ni、 Cu、 Zn、 Sr、 Y、 Zr、 Nb、 Mo、 Cd、 Sn、 Sb、 Ba、 La、 Ce、 Pr、 Nd、 Sm、 Eu、 Gd、 Tb、 Dy、 Ho、 Er、 Tm、 Yb、 Lu、 Hf、 Ta、 W、 Tl、 Pb各元素濃度均為100 mg·L-1,用2%高純硝酸逐級稀釋至各元素濃度為50 μg·L-1,此溶液為混合標準儲備液;內標元素Rh和Re儲備液濃度為100 mg·L-1,用時逐級稀釋至10 μg·L-1。
1.2 實驗步驟
1.2.1 樣品前處理 分別取25 mL 3種優級純無機酸置于干凈的Teflon溶樣杯中,120 ℃蒸干,加0.5 mL高純硝酸,再次蒸干,然后加2 mL 2%高純硝酸,加熱溶解;冷卻后轉移至5 mL離心管,添加2%高純硝酸,在電子天平上準確稱量至2.50±0.02 g,搖勻待測。樣品處理的所有過程均在桂林理工大學廣西隱伏金屬礦產勘查重點實驗室的百級超級潔凈處理臺完成。
1.2.2 ICP-MS測試 由于待測目標溶液中各元素濃度含量較低, 因此測試前將采樣錐、 截取錐、 霧化器、 霧化室和進樣系統用2%高純硝酸和超純水反復清洗, 以確保較低的本底。 另外,通過儀器設置的調諧程序, 用1 μg·L-1的7Li、59Co、89Y、140Ce、205Tl混合標準調諧液對儀器參數進行優化, 綜合考慮氧化物和雙電荷的干擾情況, 選定的最佳工作參數見表1。 此時7Li、89Y、205Tl的響應靈敏度分別為6 000±300 cps、 13 000±1 000 cps、 11 000±1 000 cps; CeO/Ce氧化物和Ce2+/Ce雙電荷產率分別為(2.10±0.10)%和(3.08±0.15)%。
分析過程中的基體效應和信號漂移等會帶來較大的誤差,一般是通過引入內標進行校正,內標元素的選擇基于以下原則:1)待測溶液中該元素的含量極低; 2)以分析元素與內標元素在等離子體中的行為相近; 3)內標元素與待測溶液中各元素的濃度接近。 因此, 選用濃度10 μg·L-1Rh和Re兩種元素的溶液為內標,通過三通在線引入, 與樣品溶液混合后進入霧化系統。 另外,由于待測樣品的體積較少(僅2.5 mL),為保證管路清洗和數據采集過程的樣品引入,所有質譜測試均采用管路較短的手動進樣模式。
2 結果與討論
2.1 測定同位素的選擇及干擾的消除
待測元素一般具有多種同位素,測定時以干擾小、豐度大為選擇原則。ICP-MS在實際測試過程中還會受到多原子、氧化物及同質異位素的干擾。本文所測樣品以2% 硝酸為介質,可能還殘留少量的氟離子、氯離子,所以進入等離子體的基體元素主要有氮、氫、氧、氟、氯等,這些元素與待測元素可能形成的干擾組分如表2所示。
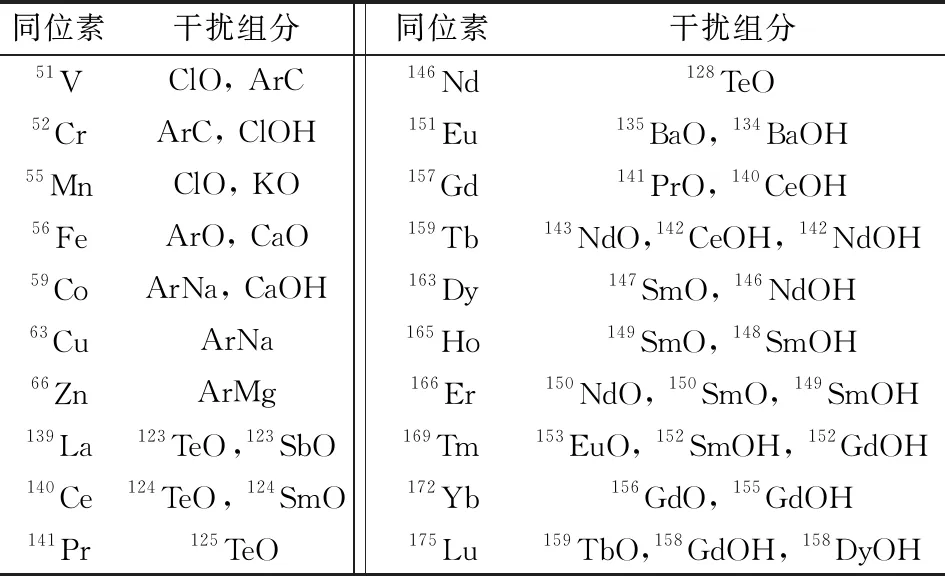
表2 多原子離子對待測元素的干擾
為了消除多原子干擾,本文采用安捷倫7500cx ICP-MS自帶的氦氣碰撞反應界面技術[12]。該技術基于動能歧視效應,即干擾粒子比同等質量的分析物離子直徑大,較大的橫截面意味著在碰撞池中與碰撞氣有更多的碰撞幾率,所以當這些多原子粒子通過碰撞池時將損失更多的動能,從而使之無法到達檢測器。
選擇參考混合標準調諧液中89Y的信號強度和CeO/Ce氧化物產率來調節碰撞反應氣的流速。在儀器工作參數設定為表1的情況下,將氦氣流速調節至6 mL·min-1,吹掃10 min后,考察89Y的信號強度和CeO/Ce氧化物產率隨碰撞氣流速的變化情況,如圖1所示。
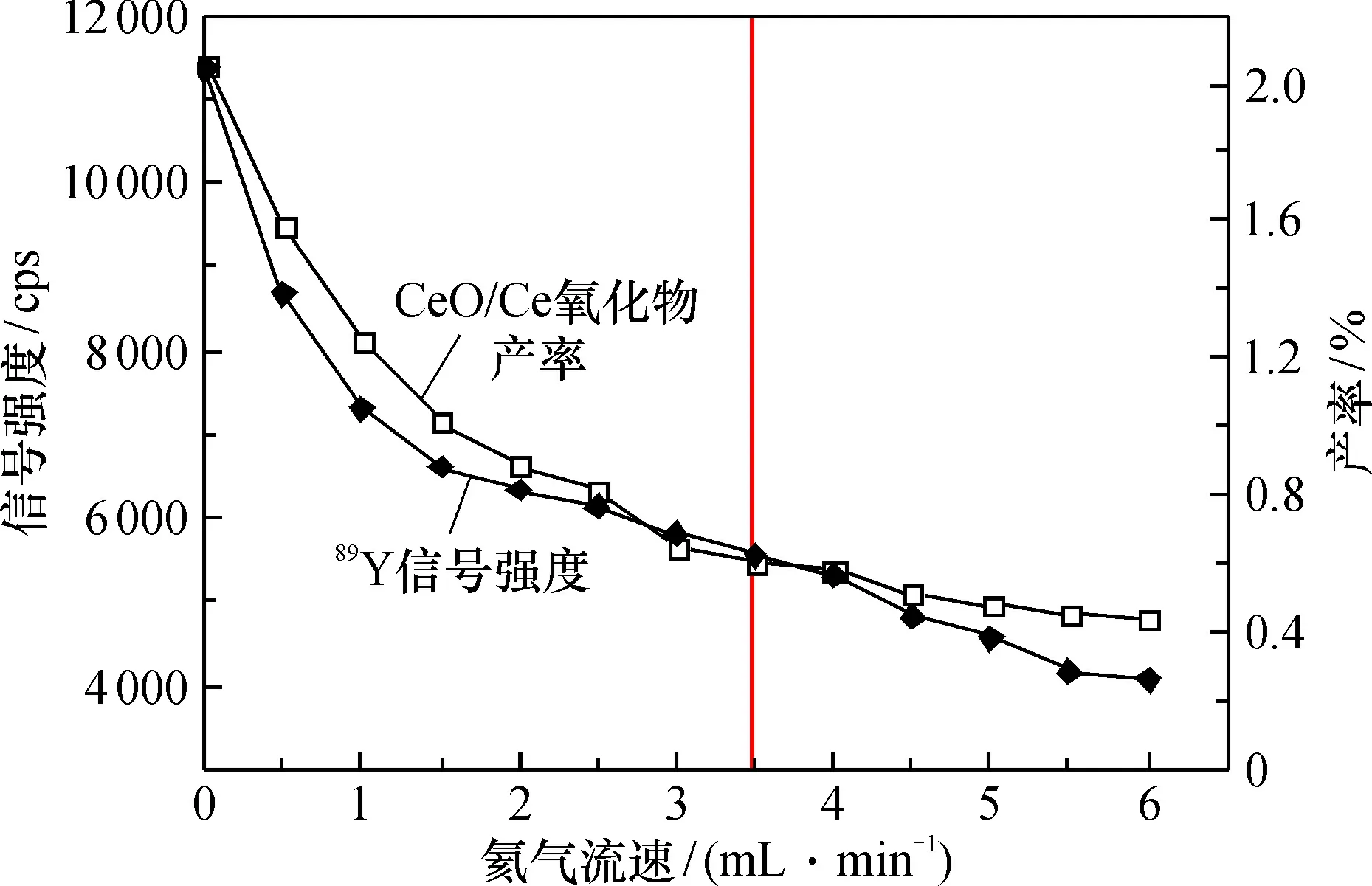
圖1 89Y的信號強度、CeO/Ce氧化物產率隨氦氣流速的變化情況
氦氣流速為0 ~ 3 mL·min-1時,89Y的信號強度和CeO/Ce氧化物產率均隨氦氣流速的增加而降幅顯著;流速繼續增加,CeO/Ce氧化物產率下降幅度變得十分緩慢,同時89Y的信號強度依然有明顯的降幅。為了確保氧化物產率始終處于較低的水平,本文選擇碰撞氣的流速為3.5 mL·min-1。
2.2 標準曲線
用2%高純硝酸將混合元素標準儲備液逐級稀釋至濃度分別為0、1.0、2.5、5.0、10.0、20.0 μg·L-1的混合元素標準溶液。在最優的實驗條件下進行測試,儀器自動生成各元素的校正曲線和相關系數,結果顯示各元素標準曲線的線性相關系數R均不低于0.999 0(表3)。
2.3 方法檢出限
方法檢出限是指能識別高于全流程空白的最低濃度,按1.2.1節流程處理6份空白樣品,在儀器工作參數設定為表1條件下對空白樣品進行測定,計算空白樣品的平均值,根據3倍信噪比和稀釋因子計算方法的檢出限,結果在0.33~20.58 ng·L-1,如表3所示。
2.4 方法準確度和精密度試驗
為了驗證該方法的可靠性,同時進行了精密度和加標回收實驗。 即添加含各元素5 ng(處理完的待測樣品中濃度預計為0.1~100 μg·L-1, 選此濃度可以與樣品更好地匹配)的標準溶液于空白樣品中,按本文的流程平行測試12份樣品,得到各元素的回收率在97.21%~103.55%,RSD在0.89%~4.96%, 說明本方法具有較好的準確度和精密度。
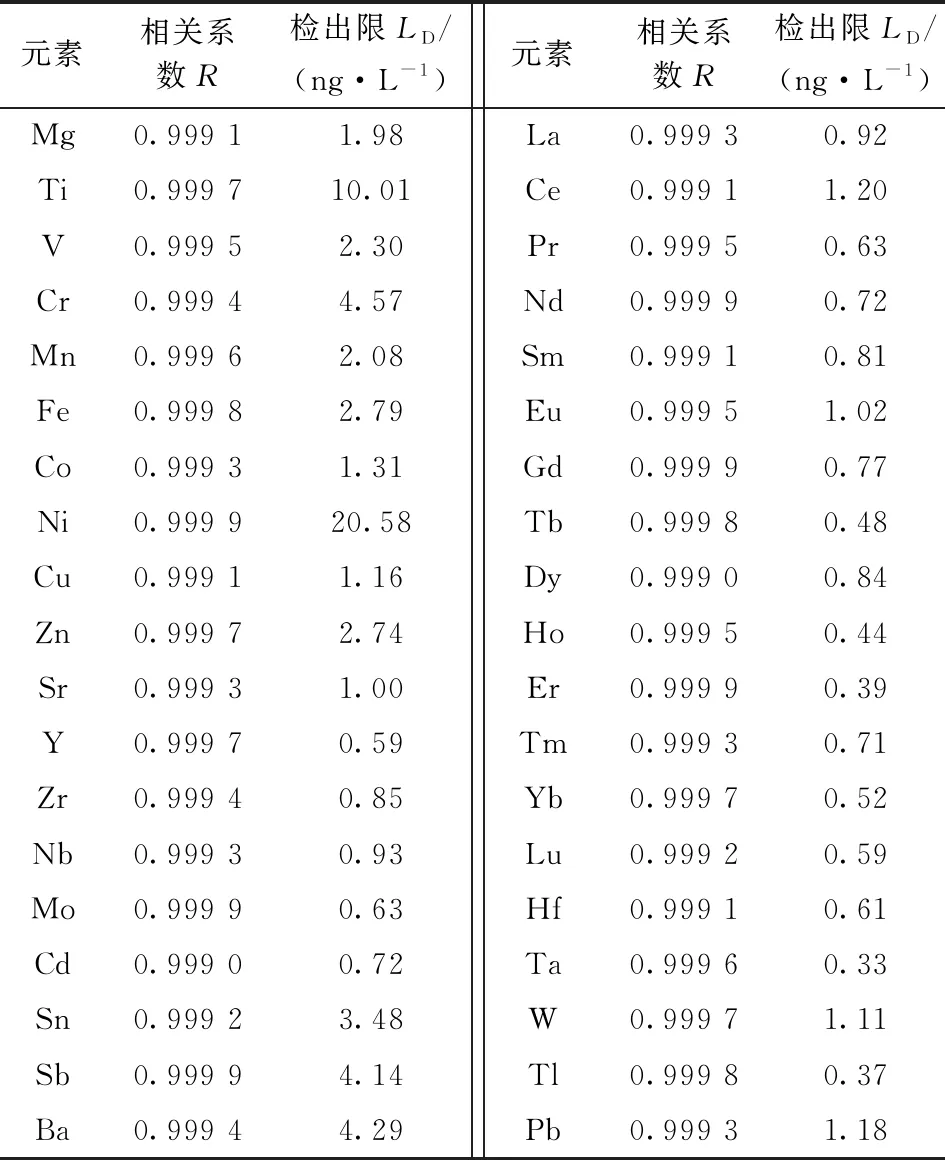
表3 各元素的檢出限及校正曲線的相關系數
2.5 樣品分析
分別對3種優級純無機酸樣品和二次純化(2 st)的氫氟酸樣品按上述實驗流程各處理6份平行樣,得到的分析結果如表4所示。在3種優級純無機酸中雜質鐵的含量普遍較高,鹽酸、硝酸和氫氟酸分別達到10.28、8.60和34.81 μg·L-1;3種酸中氫氟酸的純度相對較低,但是經亞沸蒸餾裝置純化2次后,絕大多數元素的含量均低于方法檢出限(鐵元素除外),已基本可滿足超痕量分析的工作需求。
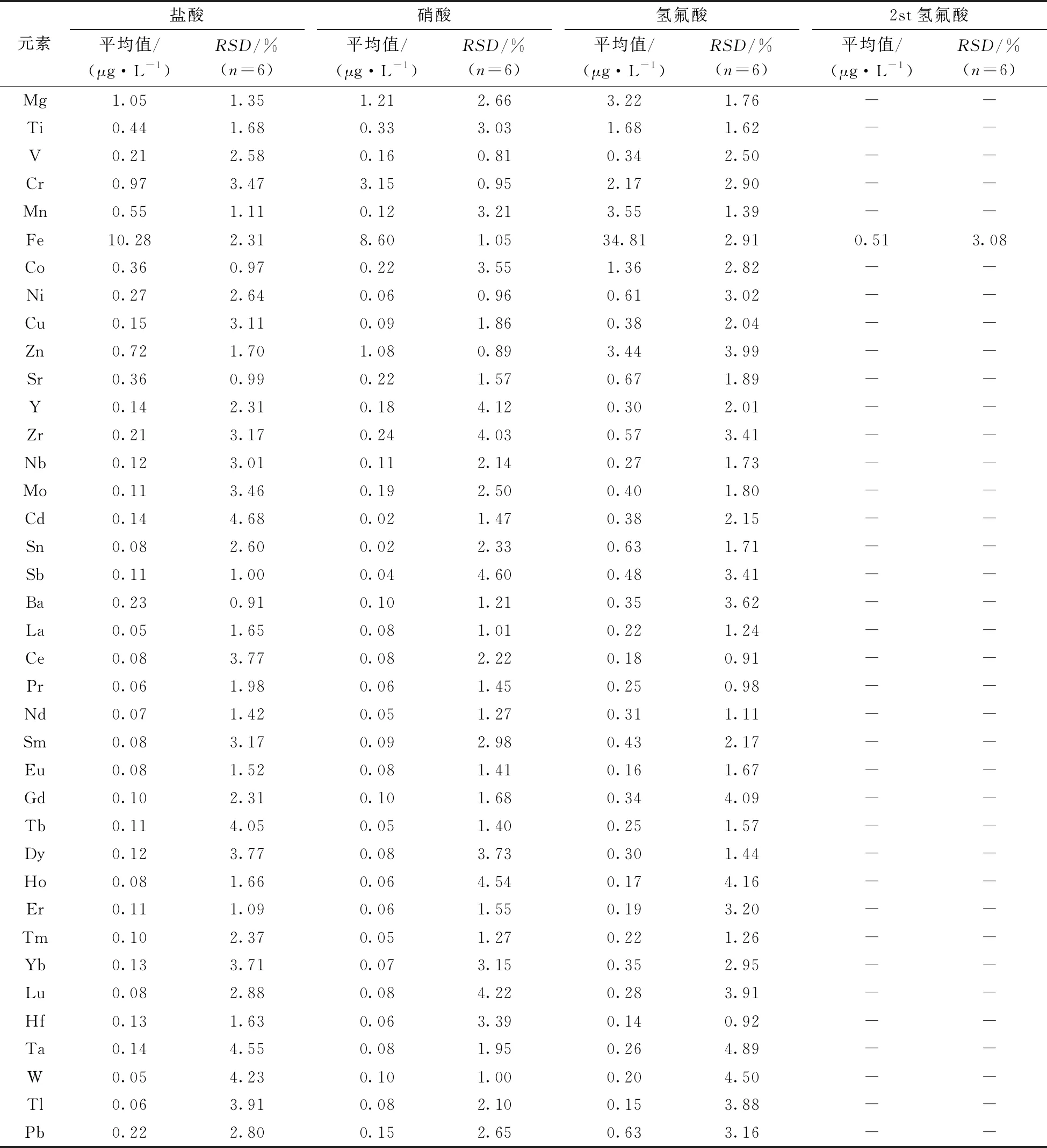
表4 樣品分析結果
3 結 論
通過對樣品的預富集和測試介質的轉換,利用帶碰撞反應池技術的電感耦合等離子體質譜儀對廣州市西隴化工有限公司生產的優級純鹽酸、 硝酸和氫氟酸中的38種痕量金屬元素進行了測定, 同時也對2次亞沸蒸餾純化過的氫氟酸中的金屬元素也進行了檢測, 結果顯示無機酸中鐵的含量普遍較高; 3種優級純無機酸以氫氟酸雜質含量最高, 而經二次純化后的氫氟酸中絕大多數的金屬離子的含量均未檢出(鐵除外)。
