抗結(jié)核藥物作用新靶點及其研究進展
2020-03-13 09:11:42陳浩武楠楠胡文輝楊忠金
中國防癆雜志 2020年3期
陳浩 武楠楠 胡文輝 楊忠金
目前,世界衛(wèi)生組織推薦治療藥物敏感性結(jié)核病使用標準四藥方案,即利福平、異煙肼、乙胺丁醇和吡嗪酰胺的6個月聯(lián)合治療(利福平、異煙肼、吡嗪酰胺和乙胺丁醇使用2個月,隨后聯(lián)用利福平、異煙肼4個月),治愈率可達到90%~95%[1-2];但是該方案的藥物及其代謝產(chǎn)物會引起肝損傷。此外,長達6個月的治療使患者的用藥依從性有所降低,如果用藥管理不完善容易導致耐藥性的產(chǎn)生。因此,我們需要時間更短的標準治療方案和能夠抵抗耐藥性菌株的新型藥物來更加快速有效地消滅結(jié)核分枝桿菌。
由于結(jié)核分枝桿菌耐藥性的越來越嚴重,世界衛(wèi)生組織和全球結(jié)核病聯(lián)盟聯(lián)合開展了抗結(jié)核新藥的開發(fā)。近10年來結(jié)核分枝桿菌分子靶點耐藥機制及其研究取得了令人振奮的成果,針對各種新靶點的抗結(jié)核新藥已經(jīng)陸續(xù)進入臨床試驗,新藥貝達喹啉和德拉馬尼已經(jīng)獲批用于治療耐多藥結(jié)核病(multi-drug resistant tuberculosis,MDR-TB)和廣泛耐藥結(jié)核病(extensively drug-resistant tuberculosis,XDR-TB)。目前各大研發(fā)機構(gòu)已經(jīng)創(chuàng)建了一個豐富的先導化合物資源庫,筆者對最有潛力的抗結(jié)核分枝桿菌新靶點和13個臨床研究階段的藥物進行梳理和分析(表1)。
一、細胞壁合成
結(jié)核分枝桿菌富含脂質(zhì)的細胞壁是新型抗結(jié)核藥物靶點的主要來源。
癸烯基磷酸基-β-D-核糖氧化酶(decaprenylphosphoryl-β-D-ribose oxidase,DprE1)是一種參與結(jié)核分枝桿菌細胞壁生物合成的關(guān)鍵酶,與癸烯基磷酸基-β-D-核糖-2-表異構(gòu)酶(decaprenylphosphoryl-β-D-ribose-2-epimerase,DprE2)一起催化D-核糖(phoryl-β-D-ribofuranose,DPR)向D-阿拉伯呋喃糖(decaprenylphosphoryl-β-D arabinofuranose,DPA)轉(zhuǎn)化。DPA是合成細胞壁阿拉伯聚糖的唯一前體。DprE1已經(jīng)成為多種化學物的作用靶點。目前正在開發(fā)的DprE1抑制劑有:苯并噻嗪酮類、氟喹諾酮類和氮雜吲哚類。其作用靶點對分枝桿菌物種具有高度選擇性,近年來DprE1正在成為開發(fā)安全和有效的抗結(jié)核藥物的研究熱點[3]。如苯并噻唑酮類化合物,它們作為還原形式的DprE1的底物進行硝基還原,產(chǎn)生亞硝基分子,特異性地攻擊活性位點半胱氨酸殘基Cys387的巰基側(cè)鏈,從而形成共價加合物并不可逆地使酶失活[4](圖1)。
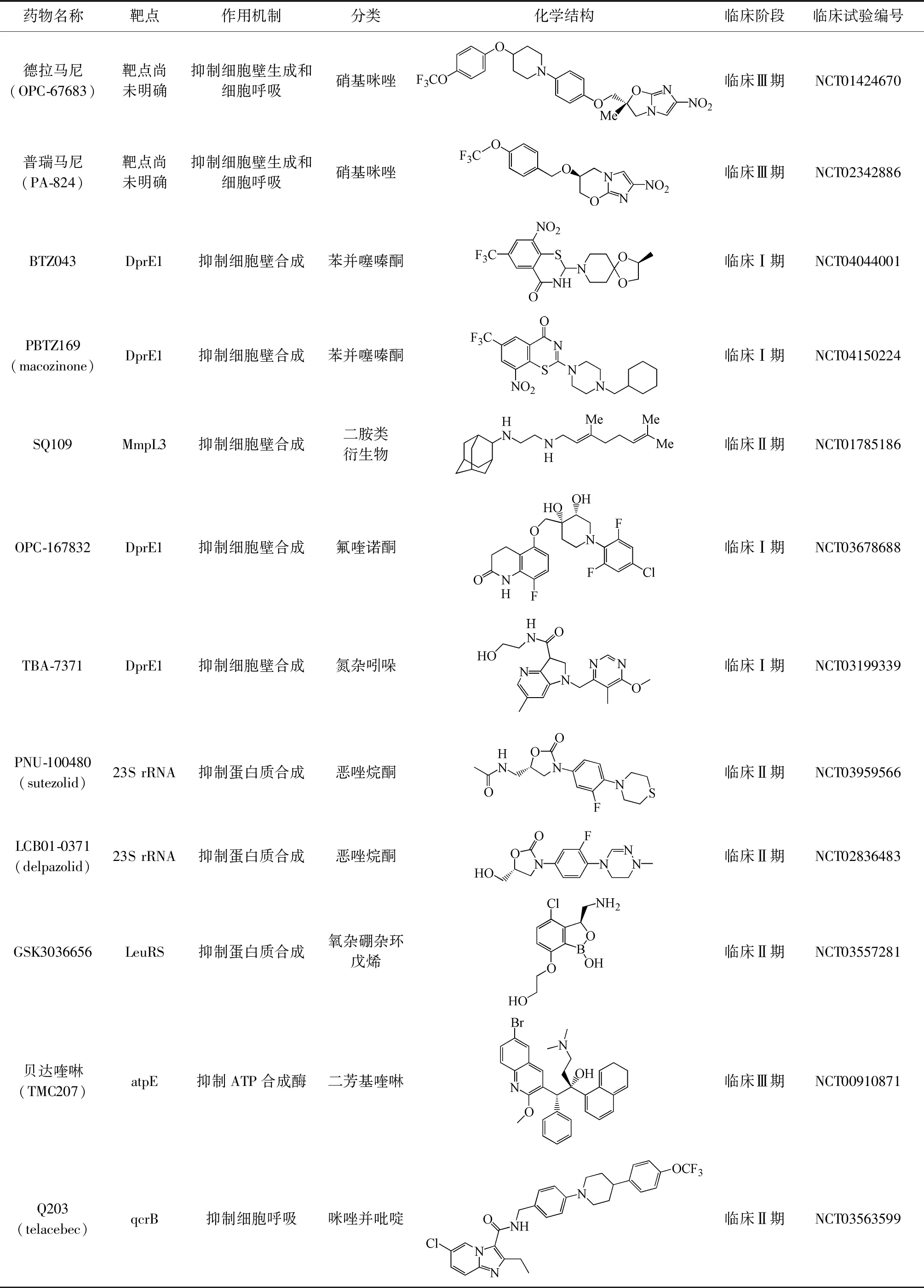
表1 抗結(jié)核新藥及其介紹
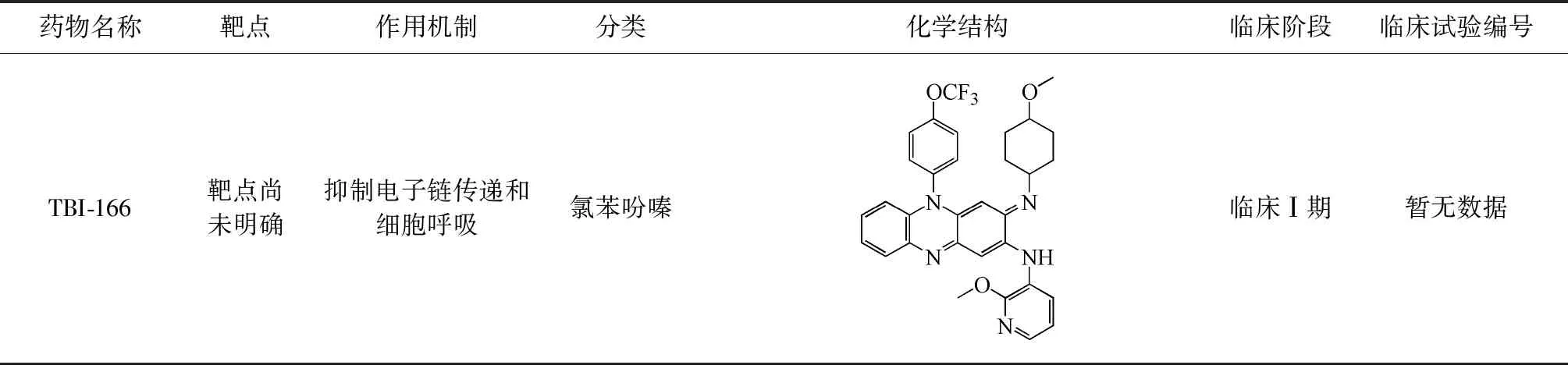
續(xù)表1
注數(shù)據(jù)來自http://www.newtbdrugs.org,截止到2020年1月
分枝桿菌膜蛋白3(mycobacterial membrane protein large 3,Mmp L3)是另一個細胞壁靶標,MmpL3作為轉(zhuǎn)移內(nèi)膜上的跨膜轉(zhuǎn)運蛋白[6],具有在體內(nèi)成為高度易受攻擊的靶標的吸引力[7]。二胺類衍生物SQ109通過作用于MmpL3進而干擾霉菌酸組裝到細菌細胞壁中而起作用,主要表現(xiàn)為抑制海藻糖二霉菌酸酯(trehalose dimycolate,TDM)的產(chǎn)生,并且霉菌酸鹽不能附著到細胞壁阿拉伯半乳聚糖上。SQ109化合物的類似物導致相似的TDM合成關(guān)閉,以及伴隨的海藻糖一霉菌酸酯(trehalose monomycolate,TMM)積累。全基因組測序顯示自發(fā)產(chǎn)生的SQ109抗性突變體都在必需的MmpL3基因中具有突變[8](圖1)。
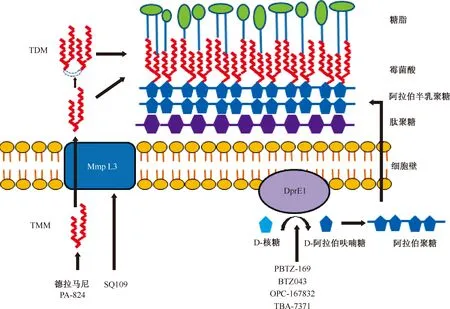
TDM:海藻糖二霉菌酸酯,trehalose dimycolate;TMM:海藻糖一霉菌酸酯,trehalose monomycolate;Mmp L3:分枝桿菌膜蛋白3,mycobacterial membrane protein large 3; DprE1: 癸烯基磷酸基-β-D-核糖氧化酶,decaprenylphosphoryl-β-D-ribose oxidas;該圖參考文獻[5]繪制圖1 抑制細胞壁合成的靶點
硝基咪唑類化合物抑制細胞壁霉菌酸合成,其分子機制尚未闡明;研究表明,硝基咪唑類化合物可抑制結(jié)核分枝桿菌細胞壁甲氧基-霉菌酸和酮-霉菌酸的合成。此外,硝基咪唑類化合物的中間體作為一氧化氮供體,引起的呼吸中毒已得到證實[9-10]。
(一)德拉馬尼(OPC-67683)
德拉馬尼是一種已經(jīng)上市的抗結(jié)核藥品,來源于硝基咪唑類化合物,具有高效的抗結(jié)核分枝桿菌活性[11]。德拉馬尼是一種前藥,其通過一種尚未知的分子機制抑制分枝桿菌細胞壁甲氧基-霉菌酸和酮-霉菌酸的合成;與異煙肼不同,該藥物不會抑制α-霉菌酸,可有效減少細菌耐藥性的形成。德拉馬尼需要通過分枝桿菌F420輔酶系統(tǒng)代謝激活才能發(fā)揮其抗結(jié)核活性[9-10]。有研究已經(jīng)證明,雙環(huán)硝基咪唑代謝途徑中的中間體作為一氧化氮供體,從而引起呼吸中毒[10]。德拉馬尼對結(jié)核分枝桿菌體外的最小抑菌濃度(minimum inhibitory concentration,MIC)范圍為0.006~0.0241 μg/ml,在體內(nèi)低劑量即可表現(xiàn)出高效的治療活性。德拉馬尼不受肝臟微粒體酶活性的影響,也不影響肝微粒體酶的活性,這表明德拉馬尼能夠與抗逆轉(zhuǎn)錄病毒藥物聯(lián)合使用,誘導或被細胞色素P450酶代謝[11]。世界衛(wèi)生組織關(guān)于德拉馬尼的使用指南中,臨床數(shù)據(jù)驗證了藥物的安全性和有效性[12]。
(二)普瑞馬尼(PA-824)
普瑞馬尼是一種雙環(huán)硝基咪唑類分子(硝基咪唑并吡喃),對復制、非復制期和缺氧條件下的結(jié)核分枝桿菌都有效。和德拉馬尼一樣,普瑞馬尼是一種前藥,其通過尚未完全闡明的分子機制抑制細胞壁霉菌酸生物合成。對普瑞馬尼作用模式的微陣列分析顯示,細胞壁抑制(如異煙肼)和呼吸中毒(如氰化物)的基因表現(xiàn)出混合效應[10]。目前,研究已經(jīng)證明普瑞馬尼直接作為一氧化氮供體,引起的呼吸中毒是普瑞馬尼抑制細菌無氧活動的關(guān)鍵因素。在缺氧非復制條件下,普瑞馬尼對呼吸復合物的作用表現(xiàn)為細胞內(nèi)ATP水平的快速下降,這與氰化物處理的現(xiàn)象相似[10]。最近,一項新的研究顯示,毒性代謝物甲基乙二醛的積累是分枝桿菌中普瑞馬尼的額外殺滅機制[13]。體外測試普瑞馬尼對藥物敏感菌株的MIC為0.015~0.25 μg/ml,對MDR-TB分離株的MIC為0.039~0.531 μg/ml;在體內(nèi)模型試驗中具有良好的活性[14]。
臨床Ⅱ期試驗數(shù)據(jù)顯示,普瑞馬尼殺菌活性的劑量范圍內(nèi)所有劑量均安全且耐受性良好。在藥物敏感性成人肺結(jié)核患者的劑量范圍研究中,普瑞馬尼被證明是安全的,耐受良好且有效[15]。在一項Ⅱ期臨床試驗的聯(lián)合用藥評估中,認為莫西沙星、PA-824和吡嗪酰胺的組合是安全的,耐受性良好,并且在治療8周期間在藥物敏感結(jié)核病中顯示出優(yōu)異的殺菌活性。該方案已準備好進入藥物敏感性結(jié)核病和MDR-TB患者的臨床Ⅲ期試驗,預期可以縮短和簡化治療[16]。
(三)BTZ043
BTZ043是一種苯并噻嗪酮類化合物,對耐多藥結(jié)核分枝桿菌耐多藥臨床分離株具有抑菌活性。BTZ043通過阻斷DprE1來抑制結(jié)核分枝桿菌細胞壁合成。BTZ043對結(jié)核分枝桿菌H37Rv的MIC為1 ng/ml,體內(nèi)試驗顯示出較低的毒性,表明BTZ043是治療結(jié)核病的有潛力的候選藥物[17]。
(四)PBTZ169 (macozinone)
PBTZ169是一種哌嗪基苯并噻嗪酮衍生物,通過BTZ043的結(jié)構(gòu)優(yōu)化得到。與BTZ043相比,PBTZ169化學合成更容易、商品成本低和藥效學更好。PBTZ169通過作用于DprE1酶抑制分枝桿菌細胞壁的合成。PBTZ169對結(jié)核分枝桿菌H37Rv的MIC為0.0625 μg/ml[18]。PBTZ169在臨床前模型中與貝達喹啉和氯法齊明具有協(xié)同作用。
(五)SQ109
SQ109是一種新型1,2-乙二胺小分子藥物,對藥物敏感和耐藥性結(jié)核分枝桿菌均具有抑菌活性。SQ109通過作用于Mmp L3靶點,干擾霉菌酸組裝到細菌細胞壁中起作用。SQ109對結(jié)核分枝桿菌H37Rv的MIC為0.015 μg/ml[19]。體外研究表明,SQ109可以增強異煙肼、利福平和貝達喹啉的抗結(jié)核分枝桿菌活性,治療患有結(jié)核病小鼠的時間縮短30%以上。SQ109可替代目前的一種或多種抗結(jié)核藥物,簡化治療,縮短目前的治療時間[20]。2017年3月,俄羅斯生物技術(shù)公司Infectex報道了SQ109安全性、有效性和耐受性方面的積極結(jié)果,Ⅱ/Ⅲ期臨床試驗結(jié)果良好,接受SQ109加標準方案治療患者的痰培養(yǎng)陰轉(zhuǎn)率為80%,明顯高于接受標準方案加安慰劑治療的患者(61%)[21]。
(六)OPC-167832
OPC-167832是一種新合成的氟喹諾酮類衍生物,其通過抑制DprE1在體外顯示出較高的抗結(jié)核分枝桿菌活性。對實驗室菌株和臨床分離菌株(包括耐藥性結(jié)核分枝桿菌),OPC-167832對結(jié)核分枝桿菌的MIC范圍為0.00024~0.002 μg/ml。OPC-167832對慢性結(jié)核病的試驗小鼠模型顯示出對細胞外和細胞內(nèi)結(jié)核分枝桿菌的有效殺菌活性,其劑量低于其他抗結(jié)核藥物。OPC-167832在體外瓊脂稀釋法和慢性結(jié)核病小鼠模型中未顯示出與其他抗結(jié)核藥物的拮抗作用[22]。對慢性結(jié)核病小鼠模型的研究顯示,德拉馬尼和OPC-167832,以及其他新的抗結(jié)核藥物聯(lián)用較標準治療方案更有效。OPC-167832與德拉馬尼聯(lián)用方案可以縮短治療時間并改善治療效果[23]。
(七)TBA-7371
TBA-7371是一種新型的氮雜吲哚類化合物,作為靶點DprE1的非共價抑制劑,TBA-7371已顯示對結(jié)核分枝桿菌的抑菌活性,MIC范圍為0.28~1.12 μg/ml,并且在結(jié)核病的嚙齒動物模型中顯示出功效[24]。
二、蛋白質(zhì)合成
抑制結(jié)核分枝桿菌的蛋白質(zhì)合成是可行的治療策略。
RNA抑制劑表現(xiàn)出一定的抗結(jié)核潛力,惡唑烷酮類化合物中利奈唑胺治療MDR-TB具有較好的效果。惡唑烷酮類化合物可以通過結(jié)合23S rRNA的V結(jié)構(gòu)域來抑制50S rRNA形成,從而阻斷蛋白質(zhì)的合成[25]。
最近研究報道了一種新的蛋白質(zhì)合成抑制劑,其靶向亮氨酰-tRNA合成酶(leucyl-tRNA synthetase,LeuRS)具有較強的抗菌活性。LeuRS是所有細胞中蛋白質(zhì)合成所需的一系列必需酶,具有2個活性位點:氨基酰化tRNA的合成位點,以及通過校對機制確定翻譯保真度的編輯位點。此外,含硼化合物被證明可通過tRNA的捕獲機制抑制LeuRS,這種機制中硼原子結(jié)合到tRNA的3′末端腺苷核苷酸的Ade76上,形成的共價化合物結(jié)合了編碼位點中tRNA的3′端,抑制亮氨酸和tRNA的合成,從而抑制蛋白質(zhì)合成。這些LeuRS抑制劑的口服生物利用度和抗菌活性,預示著這種新型藥物將得到進一步的發(fā)展[26](圖2)。
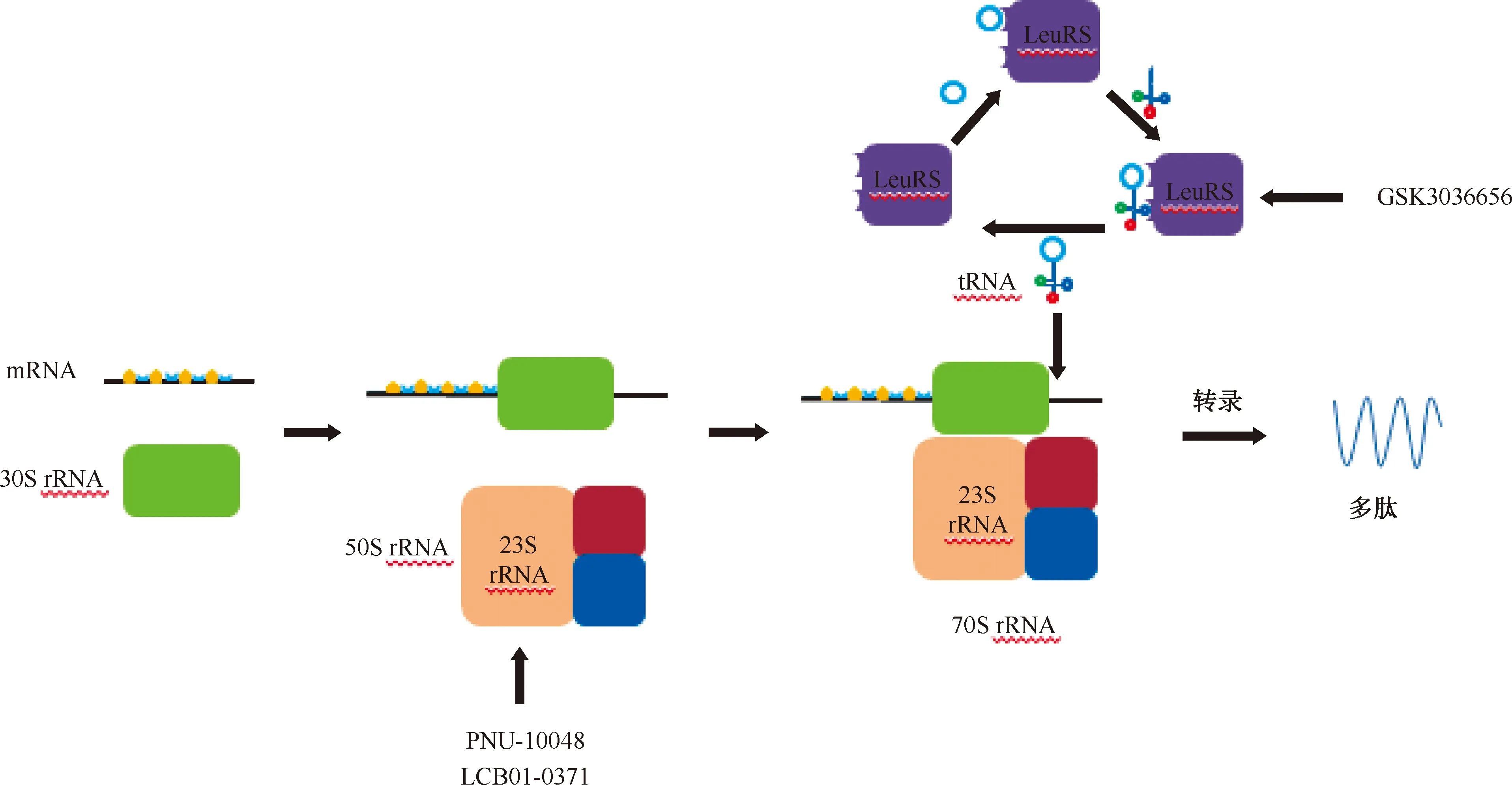
LeuRS:亮氨酰-tRNA合成酶,leucyl-tRNA synthetase;該圖參考文獻[5]繪制圖2 抑制蛋白質(zhì)合成的靶點
(一)PNU-100480(sutezolid)
PNU-100480是一種惡唑烷酮類化合物。它可以結(jié)合細菌23S rRNA的50S亞基并阻止功能性70S起始復合物的形成,從而抑制mRNA的轉(zhuǎn)錄。PNU-100480對于藥物敏感性結(jié)核分枝桿菌的MIC為0.21 μg/ml,對耐藥結(jié)核分枝桿菌的MIC范圍為≤0.0625~0.25 μg/ml[27]。Ⅰ期臨床研究結(jié)果顯示,在多次遞增劑量的安全性、耐受性、藥代動力學和藥效學評價中,PNU-100480是安全的,并且在所有測試劑量下均耐受良好[28]。
(二)LCB01-0371(delpazolid)
LCB01-0371是一種具有環(huán)狀氨基腙的新型惡唑烷酮,表現(xiàn)出較好的抗革蘭陽性菌的活性。LCB01-0371通過結(jié)合23S rRNA的V結(jié)構(gòu)域來抑制mRNA的轉(zhuǎn)錄,從而阻斷蛋白質(zhì)的形成[25]。LCB01-0371對240種多耐藥和廣泛耐藥結(jié)核分枝桿菌的90%MIC(90% minimun inhibitory concentration,MIC90)為0.5 μg/ml[29]。研究表明,LCB01-0371具有良好的安全性,由于其較好的水溶性可用于口服給藥。LCB01-0371顯示出體外和體內(nèi)抗革蘭陽性菌活性,同時具有良好的藥物代謝和藥代動力學特性[25]。
(三)GSK3036656
GSK3036656是一種新型氧雜硼雜環(huán)戊烯化合物,其靶向結(jié)核分枝桿菌的LeuRS酶可抑制蛋白質(zhì)合成。GSK3036656 對結(jié)核分枝桿菌H37Rv的MIC為0.02 μg/ml。此外,GSK3036656對結(jié)核分枝桿菌LeuRS酶具有高度選擇性(MIC=0.05 μg/ml)。GSK3036656在結(jié)核病小鼠感染模型中表現(xiàn)出良好的PK特性和抗結(jié)核分枝桿菌效果[26]。GSK3036656在南非進行的臨床Ⅱa期試驗(NCT03075410)研究表明,GSK3036656在單次和多次給藥后是安全的,耐受良好。GSK3036656藥代動力學特性和代謝產(chǎn)物圖譜使其成為一種有希望的抗結(jié)核藥,具有新的作用機制和更短的治療時間[30]。
三、能量代謝
結(jié)核分枝桿菌的喜氧性提示干擾或阻斷能量代謝可以成為有效的殺菌機制。
atpE基因編碼ATP合酶的亞單位C(質(zhì)子泵),二芳基喹啉類化合物貝達喹啉通過與結(jié)核分枝桿菌ATP合酶的亞基c結(jié)合,進而抑制ATP合成產(chǎn)生殺菌活性。這種獨特的作用方式可以減少與現(xiàn)有抗結(jié)核藥物交叉耐藥的可能性[31]。
結(jié)核分枝桿菌中的細胞色素bc1:aa3促進了對末端呼吸氧化酶抑制劑的研究。分枝桿菌細胞色素bc1:aa3由甲基萘醌、細胞色素c還原酶(bc1)和細胞色素aa3型氧化酶組成。咪唑并吡啶類化合物Q203能干擾甲基萘醌的亞基b的功能。盡管Q203對bc1:aa3復合物具有親和力,但該化合物僅具有抑菌作用。研究表明,細胞色素bd氧化酶能在Q203存在下足以維持呼吸和ATP合成,以保護結(jié)核分枝桿菌免受Q203誘導的細菌死亡。在細胞色素bd氧化酶編碼基因cydAB的遺傳缺失后,Q203完全抑制細胞呼吸作用,變成殺菌劑殺死耐受藥物的分枝桿菌,并且在體內(nèi)迅速清除了結(jié)核分枝桿菌感染[32]。這些結(jié)果表明,兩種末端呼吸氧化酶之間的相互作用可能用于抗結(jié)核藥物開發(fā)。
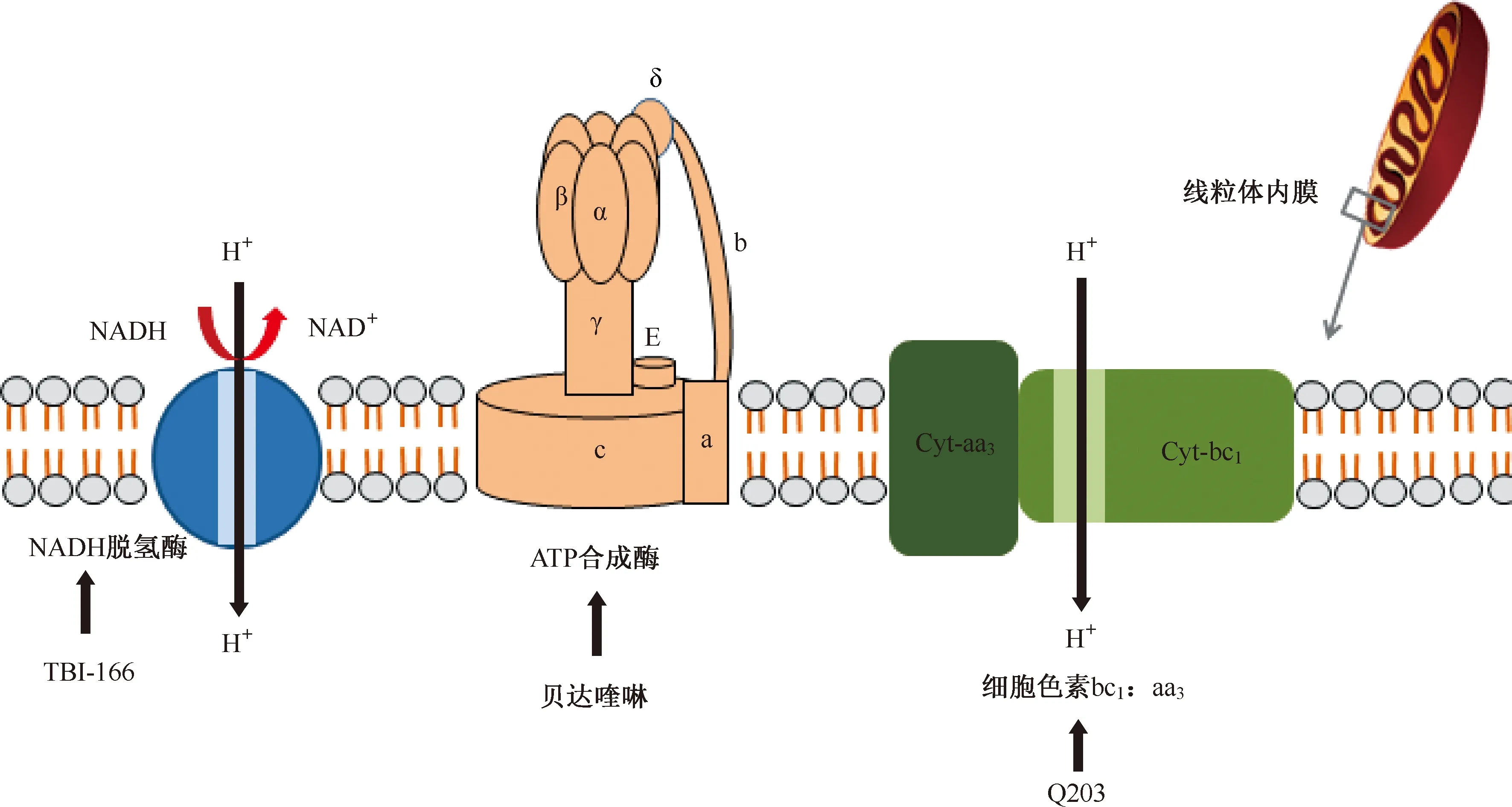
NADH:煙酰胺腺嘌呤二核苷酸,nicotinamide adenine dinucleotide;Cyt:細胞色素,cytochrome;該圖參考文獻[5]繪制圖3 抑制能量代謝的靶點
氯苯吩嗪類化合物抗結(jié)核分枝桿菌的活性極好,但其作用機制尚不完全清楚。根據(jù)研究結(jié)果推測,氯苯吩嗪類化合物可能與分枝桿菌電子傳遞鏈中的關(guān)鍵輔助因子甲基萘醌(MK-4)競爭煙酰胺腺嘌呤二核苷酸(Nicotinamide adenine dinucleotide,NADH)脫氫酶從而影響電子鏈傳遞和細胞呼吸。通過研究MK-4對氯苯吩嗪類化合物的抗結(jié)核分枝桿菌活性的影響,發(fā)現(xiàn)氯苯吩嗪類化合物和MK-4之間的競爭直接影響氯苯吩嗪類化合物對非復制和活躍生長細菌的殺滅效應,試驗中表現(xiàn)為補充MK-4阻止了藥物對非復制性細菌的活性[33](圖3)。
葛蘭素史克公司的一項研究顯示,新化合物GSK-286通過結(jié)核分枝桿菌的膽固醇分解代謝殺滅細菌,相關(guān)數(shù)據(jù)未見報道。
(一)貝達喹啉(TMC207)
貝達喹啉是二芳基喹啉類藥物的成員。貝達喹啉靶向結(jié)合結(jié)核分枝桿菌的ATP合成酶,抑制ATP合成而起到殺菌活性的作用。貝達喹啉的這種獨特作用方式最大限度地減少了與現(xiàn)有抗結(jié)核藥物交叉耐藥的可能。貝達喹啉對藥物敏感性和耐藥性分枝桿菌的MIC≤0.063 μg/ml[31]。目前正在開發(fā)的含貝達喹啉新型結(jié)核病治療方案可能會提高耐多藥結(jié)核病的治愈率。體內(nèi)外測試和臨床試驗表明,貝達喹啉具有很強的殺菌作用。
貝達喹啉在2012年獲得美國食品藥品監(jiān)督管理局批準用于治療MDR-TB,世界衛(wèi)生組織發(fā)布了使用臨時指南[34],中國于2016年已經(jīng)批準貝達喹啉用于治療MDR-TB。根據(jù)Ⅱb期臨床試驗結(jié)果,世界衛(wèi)生組織于2013年發(fā)布了關(guān)于貝達喹啉用于MDR-TB的治療手冊[35]。
(二)Q203(telacebec)
Q203是一種咪唑并吡啶化合物。Q203抑制分枝桿菌細胞色素bc1復合物的qcrB亞基,抑制細胞呼吸。Q203對臨床分離的結(jié)核分枝桿菌的50%MIC(50% minimun inhibitory concentration,MIC50)為0.84~1.56 μg/ml[32]。這些數(shù)據(jù)表明Q203是治療MDR/XDR-TB有效的臨床藥物。在慢性結(jié)核病小鼠模型中的低劑量效力及其對能量代謝的影響表明,Q203可以縮短結(jié)核病小鼠的治療時間[36]。
(三)TBI-166
TBI-166是由結(jié)核病聯(lián)盟與中國醫(yī)學科學院藥物研究所合作在抗麻風桿菌藥物氯法齊明的優(yōu)化工作中確定的新型氯苯吩嗪類化合物。TBI-166具有良好的藥代動力學特性并且沒有皮膚變色的不良反應,抗結(jié)核的療效與氯法齊明相近。TBI-166作用機制尚不完全清楚, 研究表明氯苯吩嗪類化合物可能與分枝桿菌電子傳遞鏈中的關(guān)鍵輔助因子MK-4競爭NADH脫氫酶,從而影響電子鏈傳遞和細胞呼吸[33]。TBI-166的前期研究數(shù)據(jù)表明其對結(jié)核分枝桿菌MIC為0.016 μg/ml,體外抗菌活性顯著[37]。
四、展望
近年來,抗結(jié)核新藥研發(fā)領(lǐng)域取得了重大進展。對結(jié)核分枝桿菌藥物靶點基因組學和耐藥機制的研究,使人們更深入了解了抗結(jié)核的作用靶點,這些新型靶點的發(fā)現(xiàn)給予人類對抗結(jié)核分枝桿菌以新的希望。同時,涌現(xiàn)出來的一大批新化合物結(jié)構(gòu)顯示出巨大的抗結(jié)核潛力,如BTZ043和OPC-167832 的MIC均已經(jīng)到達“ng/ml”;德拉馬尼和普瑞馬尼具有多個作用靶點,能夠有效殺傷結(jié)核分枝桿菌,降低耐藥菌株的威脅。醫(yī)藥研究工作者通過合理地對先導物進行結(jié)構(gòu)修飾,有望優(yōu)化抗結(jié)核藥物及治療方案,為患者帶來更好的療效。
