茶葉中咖啡因對有機磷類農藥殘留測定的影響
2020-04-03 05:58:40楊萍
福建茶葉 2020年3期
楊 萍
(南平市食品藥品審評與不良反應監測中心 353000)
前言:茶葉是深受人們喜愛的一種飲料這一,最近來,濫用農藥的現象也越來越嚴重,GB 2763-2016[1]也對茶葉中各種農藥殘留的限量作了具體的規定。眾所周知,茶葉富含芳香族化合物、多酚和咖啡因[2]。咖啡因等化合物會在前處理的萃取過程中與農藥殘留一起被萃取出來,如果沒有有效地去除,將會對目標農藥(毒死蜱、馬拉硫磷、殺螟硫磷)的準確定量造成影響,同時有機磷類農藥(甲胺磷、乙酰甲胺磷、氧樂果、毒死蜱、馬拉硫磷、殺螟硫磷)用氣相色譜分析時通常會一起進行凈化前處理,一起上機用FPD檢測器上機分析。這又給部分農藥殘留(甲胺磷、乙酰甲胺磷、氧樂果)的準確定量造成了失誤。
實驗儀器:島津GC-2010plus(配FPD);色譜柱:RTX-1701 30m*0.25mm,0.25um
儀器條件:進樣口溫度:220℃ 檢測器溫度:230℃
程序升溫280℃(5 min)
標準品:甲胺磷、乙酰甲胺磷、氧樂果、毒死蜱、馬拉硫磷、殺螟硫磷、咖啡因
樣品處理(方法一):依SN/T 1950-2007[3]對茶葉進行處理,同時進行加標實驗。
提取:取樣1.0g(±0.01g)于50mL塑料離心管中,加入1mL飽和氯化鈉水溶液浸泡十分鐘左右,加入15mL乙酸乙酯先均質,再加入一勺無水硫酸鈉和2勺無水硫酸鎂均質30s,用15mL乙酸乙酯洗均質頭合并提取液,蓋上蓋子振搖一會,超聲5min。把提取液和殘渣一起直接過加有2勺無水硫酸鎂的漏斗入雞心瓶中,2×5mL乙酸乙酯洗離心管,振搖,合并提取液于雞心瓶中。再用20mL乙酸乙酯沖洗漏斗上的殘渣合并洗液,35℃旋轉蒸發至剩2mL左右,待凈化。
凈化:10mL丙酮+正己烷[4](1+1,V+V)先活化TPT(10mL,2g)柱子(填料上加1cm左右高的無水硫酸鈉)下接15mL玻璃離心管,將上述大約剩2mL左右的乙酸乙酯先吸出直接過活化后的TPT柱,再用丙酮+正己烷(1+1,V+V)2.5 mL×2次洗滌雞心瓶(必要時超聲波),洗液繼續過TPT柱子,加丙酮+正己烷(1+1,V+V)繼續淋洗柱子共收集12mL淋洗液,35℃左右氮氣吹干,丙酮+正己烷(1+1,V+V)定容1mL上機測試。
混標(甲胺磷、乙酰甲胺磷、氧樂果、毒死蜱、馬拉硫磷、殺螟硫磷)與茶葉基質堆棧色譜圖,如圖1所示。
從圖1中可以看出:茶葉基質(空白茶葉)在毒死蜱、馬拉硫磷、殺螟硫磷出峰位置附近出現很大的坡(后經確認是茶葉基質中的咖啡因:前處理濃縮定容時會產生白色絮狀物,此白色絮狀物就是茶葉基質中的咖啡因析出產生的),給茶葉中毒死蜱、馬拉硫磷、殺螟硫磷準確定量帶來了嚴重影響。且氣相進了茶葉基質中的咖啡因后,要連續進十幾針的丙酮空白針,此坡才能消失,才不會對后續的出峰位置的物質定量產生干擾。
樣品處理(方法二):(目標:要去除茶葉基質中的咖咖啡因。)
提取:取樣2.0g(±0.01g)于50mL塑料離心管中,加入2mL去離子水、20mL丙酮+正己烷(1+1,V+V)溶液和一勺無水硫酸鈉,旋緊離心管蓋,渦旋1min后超聲30min,超聲期間每5min振搖一次,4000 r/min離心5min,待凈化。
凈化:移取5.0mL上清液至15mL離心管中,35℃下氮氣吹干,加入2.5mL正己烷渦旋使樣品溶解,再加入2.5mL飽和氯化鈉水溶液繼續渦旋30s說明:用飽和氯化鈉水溶液和正己烷分配可去除水溶性雜質及咖啡因)后2000 r/min離心1min,取出正己烷層,剩余溶液中加入2.5mL正己烷再提出一次。合并正己烷層過經5mL丙酮+正己烷(1+1,V+V)活化上填1cm高無水硫酸鈉的Carb/PSA柱(0.5g/6mL),用8mL丙酮+正己烷(1+1,V+V)繼續洗脫,共收集洗脫液13mL于15mL刻度玻璃離心管中,35℃水浴氮氣吹干,用正己烷定容1mL上GC-FPD檢測。同時進行加標處理(加標量:加入200ng/mL的有機磷混標1mL,與樣品同時同樣處理,相當于最終上機濃度為40ng/mL)。回收率見表1。
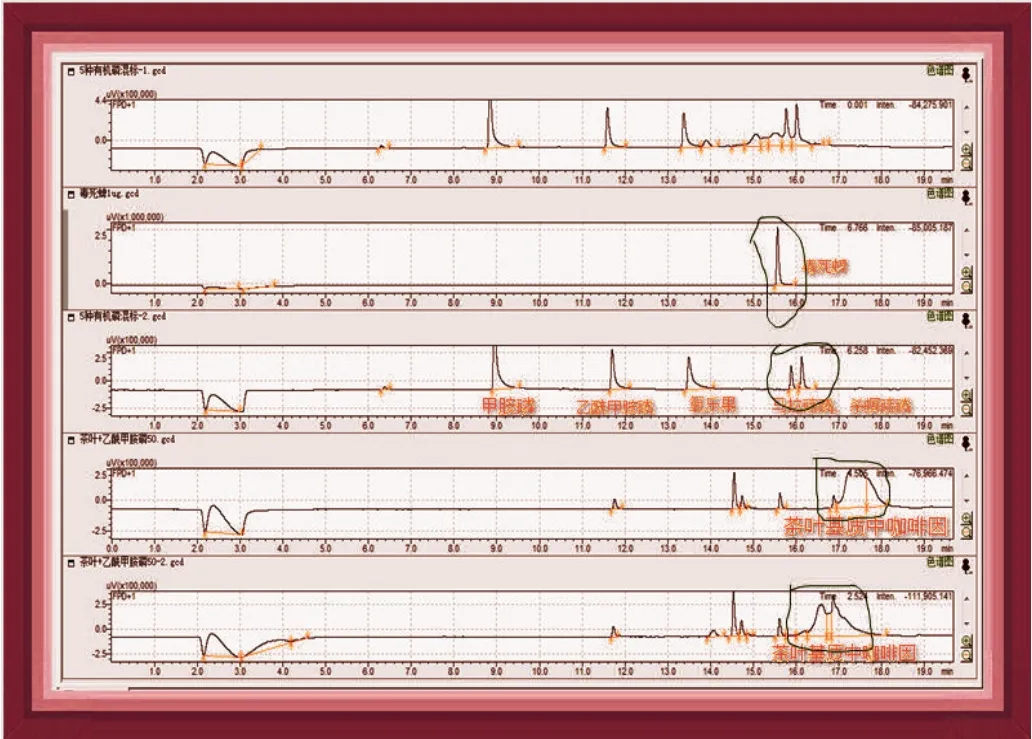
圖1 6種有機磷與茶葉基質堆棧色譜圖
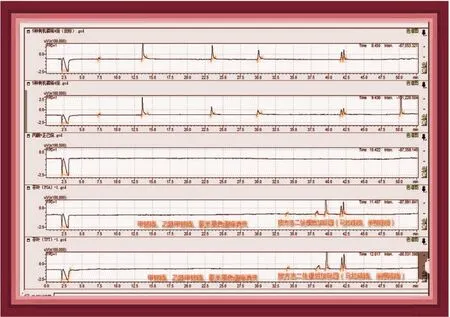
圖2 5種有機磷與茶葉基質加標(按方法二處理)堆棧色譜圖
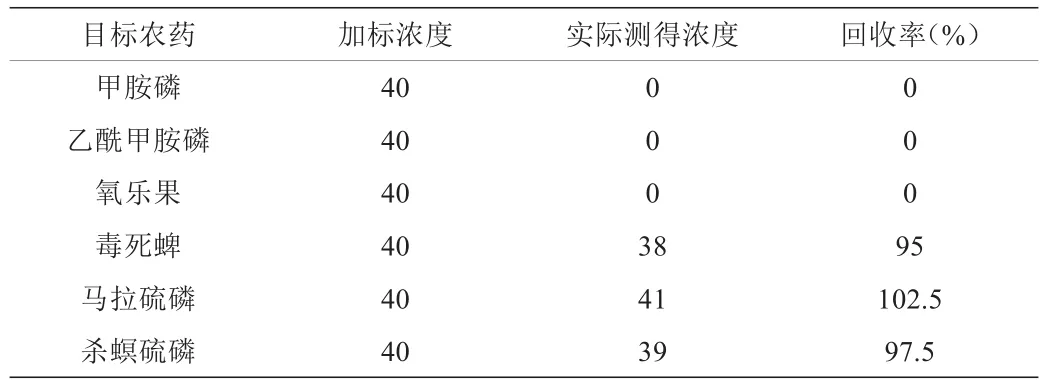
表1 用方法二處理各有機磷的回收率
從圖2及表1可以看出:使用方法二處理(用飽和氯化鈉水溶液和正己烷分配可去除水溶性雜質及咖啡因:茶葉樣品基質中的咖啡因大谷峰已消失)毒死蜱、馬拉硫磷、殺螟硫磷可以準確地給予定量了,然而甲胺磷、乙酰甲胺、氧樂果卻也同時會與咖啡因從正己烷層進入飽和的氯化鈉層而被洗脫除去。因此用方法二來處理茶葉基質的方法可以很好地去除咖啡因基質,適用于檢測毒死蜱、馬拉硫磷、殺螟硫磷,不適用于同時要檢測:甲胺磷、乙酰甲胺、氧樂果這三種有機磷的凈化方法。
(必要時)樣品處理(方法三):適用于茶葉中甲胺磷,乙酰甲胺磷的凈化方法。
取干樣0.5g于50mL塑料離心管中加2mL水,放置至少30分鐘。加入20mL乙酸乙酯和10g無水硫酸鈉,均質0.5min,用10mL乙酸乙酯清洗均質頭,合并提取液4000r/min離心5分鐘,上清液經裝有10g無水硫酸鈉的漏斗脫水于雞心瓶中,殘渣再用20mL乙酸乙酯渦漩洗滌,4000r/min離心5分鐘,上清液并入雞心瓶中,再用10mL乙酸乙酯沖洗漏斗上的殘渣合并洗液35℃濃縮至干。
凈化:用乙酸乙酯2mL×3次漩渦振蕩洗滌雞心瓶,過經5 mL乙酸乙酯活化過的上填1cm高無水硫酸鈉的硅膠柱(LC-Si 0.5g/6mL硅膠固相萃取小柱),待洗滌液流完接近硅膠柱中無水硫酸鈉頂端時,再用乙酸乙酯洗,棄去前9 mL洗液,繼續用乙酸乙酯洗并收集15mL于15mL刻度玻璃離心管中,35℃水浴氮氣吹干,用1mL乙酸乙酯(色譜純)定容,上機測試。
說明:
1)提取劑乙酸乙酯極性較強,能有效地將食品中的甲胺磷提取出來,且樣品基質中的共提取雜質相對較少;使用乙腈提取時共提取雜質稍多。使用無水硫酸鈉一方面配合均質器研磨,增加分散的均勻度,加強溶劑與樣品的接觸,提高提取效率,另一方面可以將樣品中的水分以結晶水的方式除去,既不對甲胺磷產生吸附,又避免甲胺磷溶于水導致回收率的損失。配合超聲波輔助提取,進一步提高提取效率。實驗時需先加乙酸乙酯后加無水硫酸鈉,以免無水硫酸鈉結塊導致均質困難。由于本實驗對水分的殘留較為敏感,提取時要盡可能將水分除干凈,否則影響到PSA填料的吸附性能,凈化效果變差;影響到Lc-si柱的吸附性能,可能改變柱上的洗脫規律,甚至導致實驗的失敗。可將無水硫酸鈉在650℃焙燒約4 h后備用,必要時增加用量。
作用機理研究:LC-Si柱/乙酸乙酯選擇洗脫凈化是本前處理方法的核心步驟。Lc-Si柱是經典的正相同相萃取柱,基于正相原理使雜質吸附于柱上,目標化合物隨溶劑洗出,一般使用中等偏弱極性的溶劑洗脫。乙酸乙酯是極性較強的溶劑,在這種介質中,大量中強極性及弱極性雜質均難以保留而與目標化合物一起洗出,導致凈化步驟失效。本實驗正利用了在乙酸乙酯介質中大量雜質均難以保留的特點,使其先于甲胺磷流出LC-Si柱,然后甲胺磷在特定階段流出再與仍然吸附于柱上的強極性雜質分離,達到了良好的凈化效果。
此法適用于各種復雜基質中的甲胺磷、乙酰甲胺磷的檢測(如果沒有經過LC-Si柱凈化處理且洗脫液的前9mL洗脫液要棄去,有的樣品基質會再甲胺磷、乙酰甲胺磷的出峰位置如茶葉基質中的咖啡因一樣出現很大的坡峰,給甲胺磷、乙酰甲胺磷的定量造成干擾)凈化處理。
綜合所述:茶葉中甲胺磷、乙酰甲胺磷、氧樂果、毒死蜱、馬拉硫磷、殺螟硫磷中有機磷的檢測要分成兩種不同的凈化方法進行處理。
樣品處理(方法一)適用于茶葉中甲胺磷、乙酰甲胺磷、氧樂果的凈化處理方法,因為此種凈化處理方法會同時萃取出茶葉基質中的咖啡因,茶葉富含咖啡因基質,在氣相中難以消除,會給在咖啡因出峰位置相近的化合物的定量產生干擾(如:毒死蜱、馬拉硫磷、殺螟硫磷等)
樣品處理(方法二)適用于茶葉中毒死蜱、馬拉硫磷、殺螟硫磷的測定,如果茶葉樣品沒有檢測甲胺磷、乙酰甲胺磷、氧樂果時,盡量采用此種的凈化處理方法,茶葉中的咖啡因用飽和氯化鈉水溶液和正己烷液液分配處理后,水溶性雜質、咖啡因及農藥(甲胺磷、乙酰甲胺磷、氧樂果、久效磷)會進入飽和氯化鈉水溶液層,而其它的農藥則留在正己烷層,茶葉樣品基質中的咖啡因大谷峰已消失。
樣品處理(方法三)是遇到個別復雜基質(如有次我們要檢測到含茶制品:速溶麥香紅茶)時,必要時采用的凈化處理方法,過硅膠柱(LC-Si柱)用乙酸乙酯洗脫且棄去前9mL洗脫,收集后面的15mL左右的洗脫液可保證基質中的雜質在前面9mL時洗出棄去,而目標物(甲胺磷、乙酰甲胺磷)被準確定量地收集到。
