基于卷積神經網絡的染色體交互預測算法
2020-04-08 09:30:50馬磊磊
電腦知識與技術 2020年3期
馬磊磊
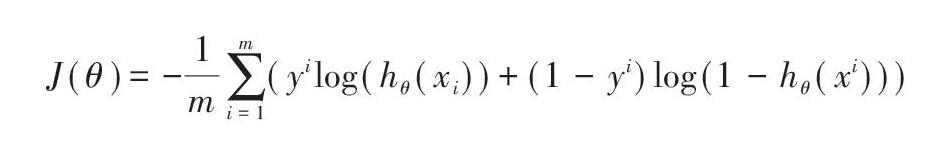
摘要:染色體是由核苷酸序列排列而成,并通過折疊盤旋形成一定的空間結構。染色體在折疊盤旋時相互接觸形成調控因子,調控生物體的各項生命活動。現階段染色體空間結構的表示方法是將三維的空間結構表示成二維的接觸矩陣,接觸矩陣中的值使用Hi-C等高通量測序技術得到,這個值表示兩個染色體片段的接觸次數。而生物方法存在實驗周期長、代價高等問題,本文針對生物實驗中存在的問題,提出了一種基于卷積神經網絡的染色體交互預測算法,并取得了較好的預測效果。
關鍵詞:染色體;空間結構;Hi-C;卷積神經網絡;染色體交互預測
中圖分類號:TP3-05 文獻標識碼:A
文章編號:1009-3044(2020)03-0198-02
1 研究背景與現狀
高通量測序技術引導的生命科學大數據時代的來臨,生物信息學相關數據出現爆炸式增長,每天產生TB級別甚至更多的序列數據,對于這些數據的挖掘和分析已經發展成為熱點的研究問題。
Hi-C技術[1]是一種以細胞核為研究對象,利用高通量測序,結合生物信息[2]分析方法,研究全基因組范圍內整個染色質DNA在空間位置上的關系,并獲得高分辨率的染色體調控元件相互作用圖譜的技術。Hi-C技術為研究染色體空間結構提供了重要的數據基礎。
Hi-C技術本質上是一種高通量測序的生物實驗方法[3],這種方法獲取染色體交互數據時,存在實驗周期長、代價高、誤差大等問題。所以使用基于計算的方法挖掘和分析相關數據,進而得到染色體交互數據具有實際意義。
現階段基于計算的染色體交互數據獲取方法主要分為兩大類:第一類基于生物實驗的Hi-C數據去偏差方法[4]。其主要通過統計計算的方法去除生物實驗中存在的偏差,得到較為精確的染色體交互數據。但是這類方法存在和生物實驗同樣的問題。第二類使用表觀修飾數據預測特定區域染色體交互[5]。其主要通過編碼表觀修飾數據,使用機器學習或者深度學習方法提取數據特征,預測特定區域染色體交互。這類方法可以較快地獲取特定區域的染色體交互,但是數據處理流程復雜、預測準確率較低,同時不能獲得所有片段的染色體交互數據。
本文針對使用表觀修飾數據預測特定區域染色體交互的方法存在的預測準確率低、預測區域具有限制性、數據處理流程復雜等問題,提出了一種以染色體親水性數據和DNA序列數據作為輸入數據,使用卷積神經網絡[6]預測全基因組染色體交互的方法。
2 實驗分析
本文以染色體親水性數據,DNA序列數據為原始的輸人數據,同時使用Hi-C實驗數據作為模型的監督數據[7]。經過數據預處理,數據編碼,特征提取等過程,進而預測全基因組染色體交互的可能性。
2.1 算法流程
本文提出的基于卷積神經網絡的染色體交互預測模型,主要使用卷積提取編碼數據的特征并用于結果預測。基于卷積神經網絡預測染色體交互方法的流程主要包括:數據預處理,數據編碼,模型設計,實驗分析四個關鍵環節。
數據預處理過程主要收集和劃分了染色體親水性數據,DNA序列數據,同時對Hi-C數據進行了數據轉化,其目的是為了生成模型預測的標簽數據。最后使用降采樣技術,對Hi-C數據生成的標簽進行了正負樣本平衡處理。
在數據編碼階段,對按照lkb長度劃分的染色體親水性數據和DNA序列數據進行了one-hot編碼。由于染色體親水性數據經過預處理轉換成8個類別,分別是0-7,所以對其進行one-hot編碼后的結果為8*1000矩陣;DNA序列數據只包含A、G、C、T四個類別,所以對其編碼后的結果為4*1000的矩陣。
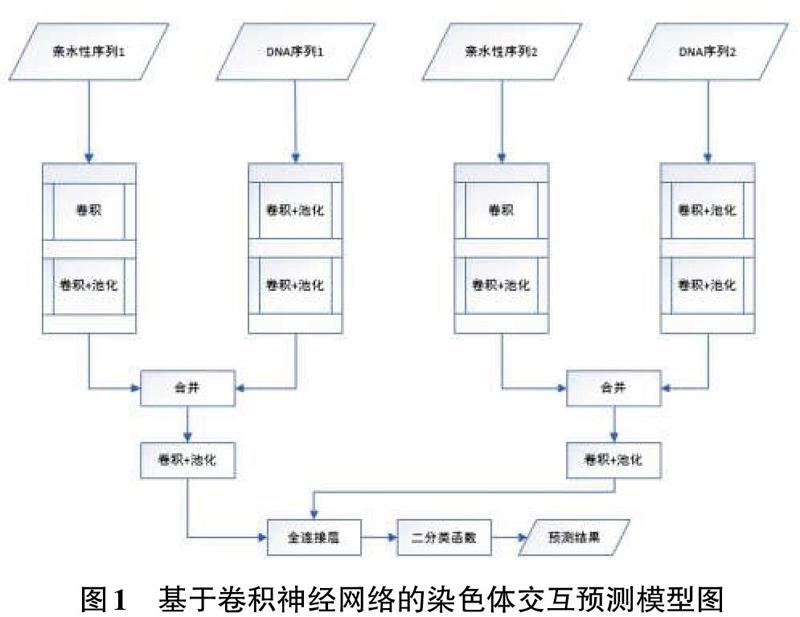
模型設計階段主要完成了基于卷積神經網絡預測染色體交互的模型設計工作。模型分別使用兩個卷積網絡獨立的提取編碼后的染色體親水性數據和DNA序列數據的數據特征,對于提取的特征進行合并,同時再使用卷積神經網絡提取更高維度的特征,最后使用全連接層加Softmax函數進行染色體交互可能性預測。
實驗分析階段主要完成了模型訓練和預測工作,同時對實驗結果進行了簡單的分析。
2.2 模型設計
卷積神經網絡具有學具表征,特征提取的能力,同時具有尺度不變性。基于卷積神經網絡預測染色體交互算法,其模型主要功能模塊包括:卷積層,最大池化層,合并層,全連接層及二分類預測層。模型的結構圖如圖1所示。
2.3 模型訓練
基于上述的染色體交互預測模型,我們使用GM12878細胞系數據作為實驗的數據集,通過降采樣方法,采樣了100W對有染色體交互的樣本記為正樣本,IOOW對沒有染色體交互的樣本作為負樣本。將采樣得到的200W對數據,按照9:1的比例劃分為訓練集和測試集,訓練集180W對,測試集20W對。
由于模型是一個二分類模型,模型的正樣本標簽為l,負樣本標簽為0。在模型訓練過程中,文中選取了交叉熵損失函數[8],使用梯度下降技術進行訓練,同時使用Adam算子進行優化。模型的損失函數如下所示:
2.4 模型預測及結果分析
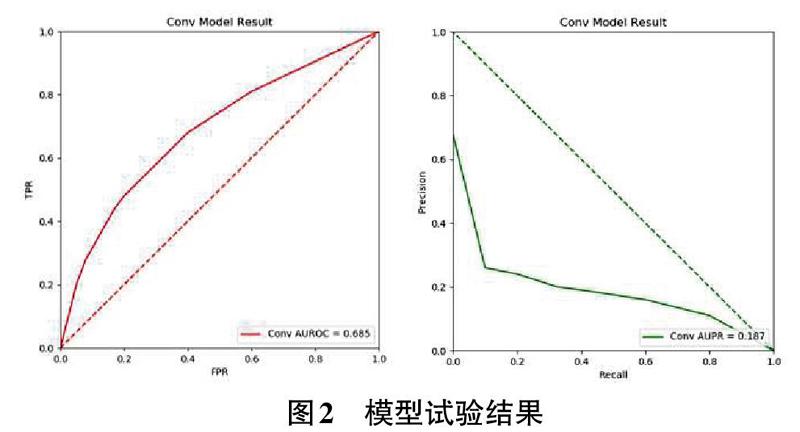
基于上述的訓練模型,我們使用20W對測試集對模型訓練效果進行了測試和評價。考慮到模型是一個二分類模型,文中使用了AUROC和AUPR作為模型的評價指標。AUROC指ROC曲線下的面積,其意義表示隨機獲取一個正樣本和一個負樣本,模型預測為正的結果把正樣本排在負樣本之前的概率,可以有效地衡量一個二分類模型的效果。AUPR指Recall值和preclsion值形成的曲線下面積。我們使用CM12878細胞系測試數據集的測試結果如圖2。
觀察模型的預測結果,我們發現,本文提出的基于卷積神經網絡的染色體交互預測模型在進行染色體交互可能性預測時AUC值可以達到0.685,同時預測結果的PR曲線下面積也可以達到0.187。
3 結論
本文分析了染色體交互可能的形成機制[9],以染色體親水性數據,DNA序列數據作為模型的基礎輸入數據,使用基于卷積神經網絡的深度學習模型預測了染色體交互的可能性。實驗結果表明,基于卷積神經網絡的染色體交互預測模型可以有效地預測全基因組范圍內lkb長度的染色體交互,同時可以得到較高的AUC值,本模型對于使用深度網絡模型預測染色體交互具有很好的實用價值和參考意義。
參考文獻:
[1] JinF, Li Y.Dixon J R.et al.A high-resolution map of thethree-dimensional chromatin interactome in human cells[J].Nature, 2009,503(7475):290-294.
[2]董建成霍奇曼,林安華,生物信息學[M].北京:科學出版社,2010.
[3]呂紅強,郝樂樂,劉源,等.基于生物信息學的Hi-C研究現狀與發展趨勢(三維基因組專刊稿件)[J].遺傳,2019: 0-0.
[4] Lettice L A.Disruption of a long-range cis-acting regulatorfor Shh causes preaxial polydactyly[J]. Proc. Natl Acad. Sci.USA 99, 2002: 7548-7553.
[5] Wenran L,Hung W W, Rui J.DeepTACT: predicting 3D chro-matin contacts via bootstrapping deep learning[J]. Nucleic Ac-ids Research. 2018 (10):10.
[6] S.Chopra, R.Hadsell, and Y.LeCun. Leaming a similarity met-ric discriminatively, with application to face verification, inProc[J]. IEEE Comput. Soc. Conf. CVPR, 2002:539-546.
[7] Rao,S.S.et al.A 3D map of the human genome at kilobaseresolution reveals principles of chromatin looping[J]. Cell. 2014 (159):1665-1680.
[8]閻平凡,張長水.人工神經網絡與模擬進化計算[M].北京:清華大學出版社,1900.
[9]吳燕如,珠杰,管美靜,基于神經網絡的目標檢測技術研究綜述及應用[J].電腦知識與技術,2019,15(33):181-184.
