固相萃取小柱-超高效液相串聯質譜測定牛奶中氯霉素
2020-04-25 12:18:42馬曉年梁志堅李怡
中國乳品工業 2020年3期
馬曉年,梁志堅,李怡
(昆明市疾病預防控制中心,昆明650228)
0 引 言
牛奶中含有豐富的蛋白質、脂肪、維生素和礦物質等營養物質。2008 年1 月15 日,衛生部發布的《中國居民膳食指南(2007)》介紹了新版《膳食寶塔》凸顯牛奶價值,牛奶是攝取營養元素的催化劑以及健康的關鍵,是健康膳食的基石[1]。但由于奶牛飼養過程中,對泌乳期奶牛用藥不當、違反國家規定把抗生素作為奶牛飼料添加劑使用、未經徹底清洗和消毒患病奶牛使用過的擠奶工具及貯奶設備、高溫季節摻雜抗生素防止牛奶酸敗等原因,造成牛奶抗生素污染[2-3]。牛奶中的抗生素殘留主要以氯霉素為主,氯霉素(Chloram?phnicol,CAP)屬廣譜抗生素,能抑制細菌蛋白質的形成,廣泛用于動物各種傳染性疾病的治療,特別是用于奶牛乳腺炎的治療,從而易使奶及奶制品中存在氯霉素殘留。由于氯霉素可引起再生障礙性貧血和粒狀白細胞缺乏癥等疾病[4],1999 年9 月13 日中華人民共和國農業部發布了《動物性食品中獸藥最高殘留限量》[5]的通知,規定了氯霉素在所有食品動物的可食用組織不得檢出。
目前,氯霉素主要通過固相萃取小柱進行前處理的提取、凈化[6-7],測定方法有液相色譜[8]、液相色譜-串聯質譜[9-15]。液相色譜采用保留時間定性,易出現假陽性,而液相色譜-串聯質譜采用核質比定性,具有更高的準確度。液相色譜-串聯質譜較于液相色譜具有更高的靈敏度,實現對痕量物質更低的檢出限。前處理部分,國標方法[11]采用C18 固相萃取小柱對牛奶進行提取、凈化,風險監測工作手冊[12]采用MSC 固相萃取小柱提取、凈化雞蛋等高蛋白樣品。依據上述信息,本實驗對比了C18、MSC 兩種固相萃取小柱對牛奶樣品中氯霉素的提取效果,為牛奶中氯霉素的測定提供參考依據。
1 材料與方法
1.1 儀器與試劑
QTRAP 4500 質譜分析儀(美國AB SCIEX);Ag?ilent 1290 高效液相色譜儀(美國安捷倫);高速冷凍離心機(湖南赫西儀器);振蕩器(常州金壇恒豐儀器制造有限公司);旋轉蒸發儀(德國IKA);氮吹儀(Reeko Auto EVA-30);電子天平(Sartorius,0.01 mg);均質器;Agela Technologies Cleanert MCS-SPE固相萃取小柱(500 mg/6 mL);Agela Technologies Cleanert S C18-SPE固相萃取小柱(500 mg/6 mL)。
液態牛奶,市購;氯霉素(壇墨質檢-國家標準物質中心);氯霉素-D5(壇墨質檢-國家標準物質中心);甲醇、乙酸乙酯(色譜純,美國JT Baker);氨水、氯化鈉(分析純);正己烷(優級純);實驗用水為超純水。
1.2 方法
1.2.1 標準溶液配制
準確量取氯霉素標準溶液25 μL,用甲醇稀釋并定容至100 mL,得到25.00 ng/mL 的氯霉素應用液。準確量取氯霉素-D5 標準溶液20 μL,用甲醇稀釋并定容至100 mL,得到20.00 ng/mL 的氯霉素-D5 應用液。標準應用液均于4 ℃條件下保存。
1.2.2 樣品的提取
取10 g(精確至0.01g)均質好的牛奶樣品于50 mL離心管中,加氯霉素-D5 標準應用液100 μL,再加乙酸乙酯20 mL,振蕩15 min,5 000 r/min 離心10 min,取乙酸乙酯層于雞心瓶中。再加20 mL 乙酸乙酯重復提取一次,合并兩次提取液,于40 ℃水浴旋轉蒸發至干。用5 mL 4%氯化鈉溶液溶解殘留物,加5 mL 正己烷振蕩混合1 min,靜置分層,棄去正己烷。再加正己烷5 mL,重復提取一次,取下層液備用。
1.2.3 樣品的凈化
C18及MCS小柱依次用5 mL甲醇、5 mL水活化,取備用液分別過C18 及MCS 固相萃取小柱,用5 mL水淋洗抽干,再用5 mL 甲醇洗脫,收集洗脫液于40 ℃下氮氣吹干。用1.0 mL甲醇溶解殘留物,渦旋混勻,過0.22 μm 濾膜,供超高效液相-串聯質譜(UPLC-MS/MS)測定。
1.2.4 儀器條件
色譜:Syncronis C18 色譜柱(100×2.1 mm,1.7 μm);流動相A:0.05%氨水,流動相B:乙腈,梯度洗脫程序件(見表1);流速:0.40 mL/min,分析時間5 min,進樣體積5 μL,柱溫30 ℃。
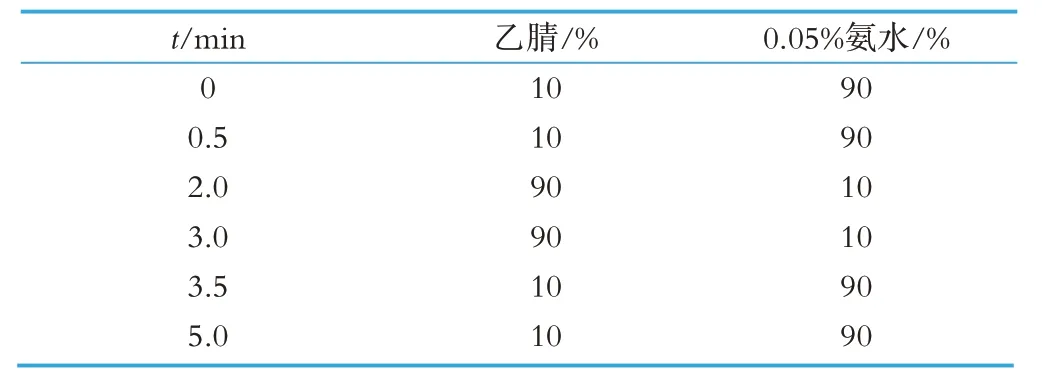
表1 流動相梯度洗脫
質譜:離子源:電噴霧離子源(ESI);掃描方式:負離子模式;檢測方式:多反應監測(MRM);電離電壓:4.0 kV;檢測方式:多反應檢測(MRM);離子源溫度:500 ℃;噴霧器壓力:50 psi;輔助加熱氣:50 psi;氣簾氣壓力:35 psi;其他參數見表2。
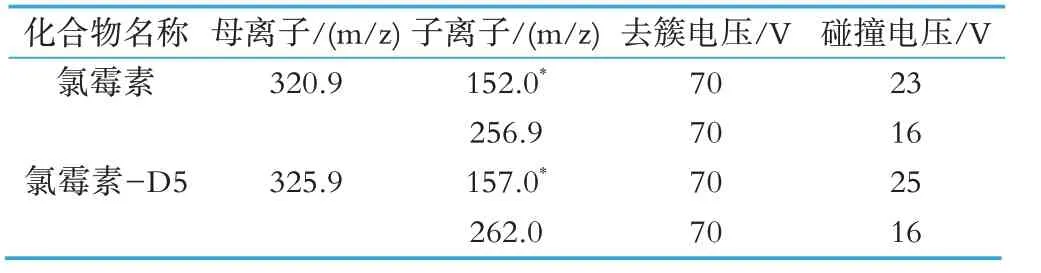
表2 各化合物質譜參數
2 結果與分析
2.1 質譜條件優化
根據待測物的性質,使用氯霉素及氯霉素-D5 標準應用液在ESI-模式下分別進行質譜條件優化。選擇每種藥物響應強度最高的m/z 值作為母離子,進行子離子掃描,并優化去簇電壓和碰撞電壓。并在MRM 模式下優化了氣簾氣、離子源溫度、噴霧氣、輔助加熱氣。
2.2 色譜條件優化
本文實驗了Hypersll GOLD色譜柱(100×2.1 mm,1.9 μm),Syncronis C18 色譜柱(100×2.1 mm,1.7 μm),發現采用Hypersll GOLD 色譜柱(100×2.1 mm,1.9 μm)分離時,色譜峰展寬,響應值偏低。采用Syncronis C18色譜柱(100×2.1 mm,1.7 μm)分離時,色譜峰得到較大改善,峰形收窄,響應增高,基線噪音降低,實現更低檢出限。
本文選擇流動相時考慮到是ESI-模式,所以使用了0.05%氨水及具有改善峰形作用的乙腈。低流速時,峰形展寬、拖尾,綜合分離效果及柱壓等因素,最終選擇0.4 mL/min 流速。圖1 為1.00 ng/mL 的氯霉素標準溶液MRM 譜圖。

圖1 CPA標準MRM譜圖
2.3 樣品前處理方法的優化
2.3.1 提取溶劑的選擇
本文試驗了乙腈-乙酸乙酯(10:90)、乙腈-乙酸乙酯(20:80)、乙腈-乙酸乙酯(50:50)及乙酸乙酯的提取效果。乙腈-乙酸乙酯(10:90)、乙腈-乙酸乙酯(20:80)分層效果不理想,震搖乳化。乙腈-乙酸乙酯(50:50)提取,分層效果較好(分為乙腈層、磷脂層、乙酸乙酯層),但加標回收率較低,在21.6%~48.5%之間。乙酸乙酯,振蕩提取效果較好,回收率均能達50%以上。
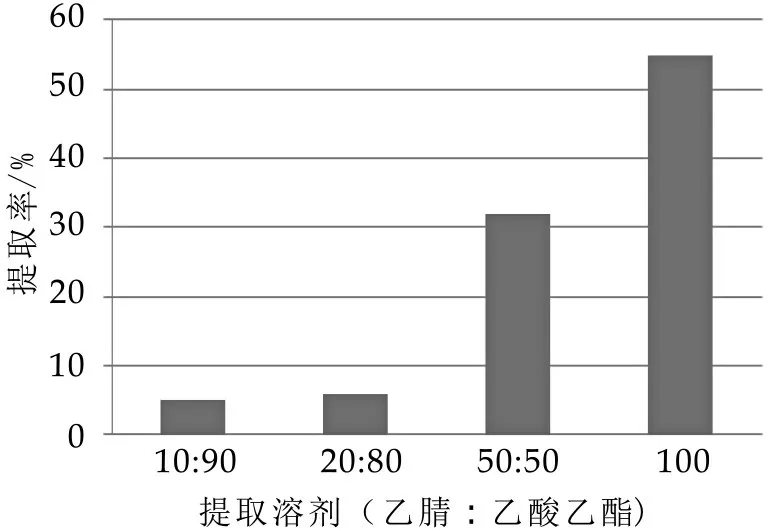
圖2 不同提取溶劑對比
2.3.2 提取方法的選擇
本文比較了《2019 年國家食品污染物和有害因素風險監測工作手冊》[11]中用乙酸乙酯提取一次,氮氣吹干水溶解過固相萃取小柱提取、凈化,及GB 29688-2013《牛奶中氯霉素殘留量的測定液相色譜-串聯質譜法》[12]中乙酸乙酯提取兩次,旋轉蒸發至干,水溶液溶解過固相萃取小柱提取、凈化。實驗發現,手冊的提取方法損失較大,回收率均于60%以下,國標方法提取效果較佳。
2.3.3 固相萃取柱的選擇
本文進行了0.10、0.50、2.50、10.00 ng 4 個水平的加標回收實驗,比較了C18 及MCS 兩種固相萃取小柱對氯霉素的保留能力。結果表明MCS 固相萃取小柱4 個水平的回收均實現75%以上,而C18 固相萃取小柱在低水平回收實驗中效果不佳,回收率低于50%。實驗結果見表3。
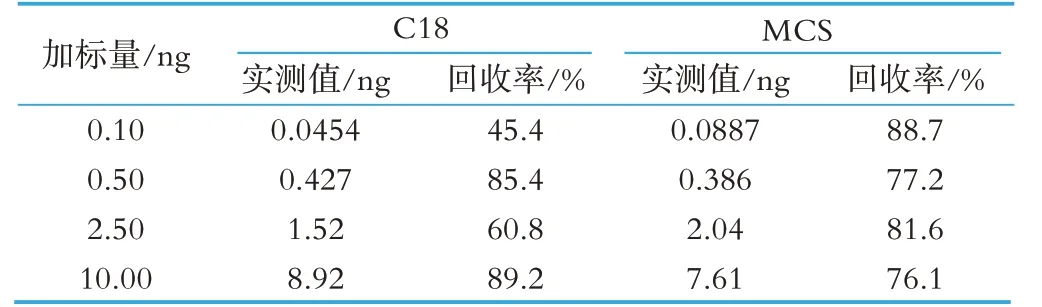
表3 兩種固相萃取柱的回收率(%)
2.4 標準曲線及檢出限
用甲醇將氯霉素-D5 及氯霉素標準應用液制備成含氯霉素-D5 2.00 ng/mL,含氯霉素分別為0.01、0.05、0.10、0.50、1.00、2.50、5.00、10.00 ng/mL 的標準工作溶液,在確定的分析條件下進行測定,以標準溶液質量濃度為橫坐標,氯霉素與氯霉素-D5 響應值之比為縱坐標繪制標準曲線,得到的線性方程、相關系數及檢出限見表4。

表4 氯霉素的線性方程、相關系數、檢出限
2.5 精密度及準確度
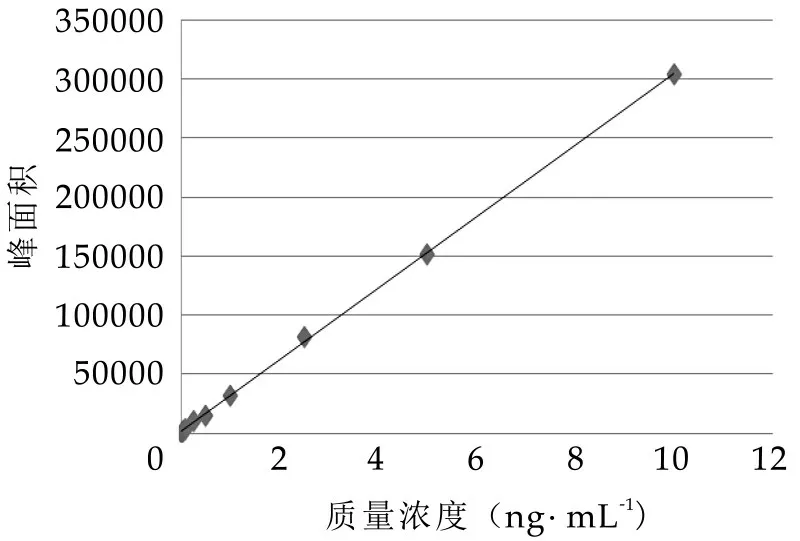
圖3 工作曲線
取不含氯霉素的牛奶樣品分別過C18 及MCS 固相萃取小柱進行三水平加標回收率實驗,結果見表5。由表中可見,C18 固相萃取小柱回收率在31.4%~155%之間,在低水平及高水平的回收率均不理想,而MCS 固相萃取小柱在三個水平的加標回收率均能達到50.4%以上。結合上述固相萃取小柱回收試驗,說明MCS 固相萃取小柱有更良好的準確性,適宜于牛奶中氯霉素殘留的痕量測定。圖4 為牛奶樣品添加氯霉素的MRM 譜圖。
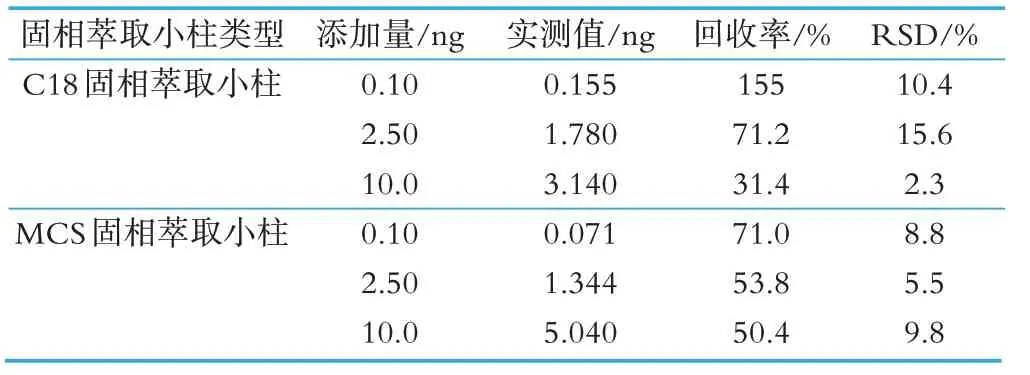
表5 氯霉素在不同加標水平下的加標回收率及相對標準偏差(n=3)

圖4 加標樣品MRM譜圖
3 結 論
本研究采用2 種不同的固相萃取小柱對牛奶中的氯霉素進行提取、富集和凈化,通過對回收率、精密度、定量限等指標進行考察,得出MCS 固相萃取小柱損失小,回收率高,同時檢出限可達到0.004 μg/kg,遠低于國標方法,且穩定性高,適用于大量樣品的集中測定。
建立了UPLC-MS/MS 內標法測定牛奶中氯霉素含量的方法。分析過程中引入氯霉素-D5 作為內標物,對于分析全過程中導致目標物損失給予完全的校正,改善了方法的精密度。以Syncronis C18 色譜柱分離,流動相為0.05%氨水和乙腈,電噴霧負離子MRM 模式檢測。該法測定周期短、定性、定量準確,適用于痕量氯霉素的檢測。
