電感耦合等離子體發(fā)射光譜法-標(biāo)準(zhǔn)加入法測(cè)定磷礦中鉛鎘含量
2020-05-15 10:14:58伍斯靜鄔景榮吳雪英梁菲萍韋偉平
化工設(shè)計(jì)通訊 2020年4期
伍斯靜,鄔景榮,吳雪英,梁菲萍,韋偉平
(1.中國(guó)檢驗(yàn)認(rèn)證集團(tuán)廣西有限公司,廣西防城港 538001;
2.廣西中檢檢測(cè)技術(shù)服務(wù)有限公司,廣西防城港 538001)
云南和貴州是我國(guó)磷礦資源最為豐富的兩個(gè)省份,一直以來(lái),從云南和貴州出口至日本的磷礦石都是通過(guò)鐵路運(yùn)輸?shù)奖辈繛掣郏缓笱b船出口到日本。日本對(duì)產(chǎn)品質(zhì)量和檢驗(yàn)檢測(cè)結(jié)果準(zhǔn)確性要求非常高,每一批貨物在出口前均要求進(jìn)行裝船前預(yù)檢驗(yàn)和裝船檢驗(yàn),貨物到達(dá)日本卸貨港后,也需再次進(jìn)行卸貨檢驗(yàn),且需多方同時(shí)進(jìn)行檢驗(yàn)分析比對(duì)結(jié)果,一旦超出允差范圍,將會(huì)遭到日方對(duì)我國(guó)檢驗(yàn)檢測(cè)技術(shù)的質(zhì)疑,嚴(yán)重影響各方合作關(guān)系。
由于磷礦中Pb、Cd 等重金屬元素會(huì)污染土壤、大氣及水等生態(tài)環(huán)境,對(duì)動(dòng)物、植物生長(zhǎng)發(fā)育有害;用于生產(chǎn)磷肥、磷酸等化工品生產(chǎn)時(shí),將會(huì)引入有害元素,影響產(chǎn)品質(zhì)量。因此磷礦中Pb、Cd 等重金屬元素是出口日本的必檢項(xiàng)目。但由于日本對(duì)產(chǎn)品質(zhì)量要求高,一般出口日本的磷礦產(chǎn)品,鉛鎘含量指標(biāo)要求非常低,一般要求鉛含量小于10mg/kg,鎘含量小于1.0mg/kg,目前很多檢測(cè)方法和設(shè)備均無(wú)法滿足要求,給檢測(cè)分析帶來(lái)了一定難度。
目前,我國(guó)對(duì)于磷礦中鉛、鎘的檢測(cè)方法,主要有GB/T 29875—2013《 磷礦石和磷精礦中鉛、砷、汞含量的測(cè)定》(適用于鉛含量>0.001 0%)和GB/T13551—1995《磷礦石和磷精礦中氧化鎘含量的測(cè)定 火焰原子吸收光譜法》(適用于氧化鎘含量大于0.000 1%),均采用火焰原子吸收法,標(biāo)準(zhǔn)適用范圍達(dá)不到要求,且標(biāo)準(zhǔn)忽略了磷礦中共存組分對(duì)測(cè)定元素的干擾,特別是測(cè)定元素含量較低時(shí),嚴(yán)重影響其準(zhǔn)確度。
而日方采用新日本檢定協(xié)會(huì)制定的方法,其中測(cè)定Cd 的樣品前處理方法(王水溶樣)與我國(guó)國(guó)標(biāo)方法基本相同,但測(cè)定方法有所不同,日方采用的是無(wú)火焰原子吸收光譜法,我國(guó)國(guó)標(biāo)采用的是火焰原子吸收光譜法,日方方法達(dá)到更低的檢出限。但無(wú)火焰法通常是石墨爐法,此方法穩(wěn)定性較差,且石墨管消耗成本大。而測(cè)定Pb 則采用了有機(jī)溶劑萃取-火焰原子吸收光譜法,與我國(guó)的測(cè)定結(jié)果存在差異。日方的方法雖消除了磷礦中P 和Ca 等共存大量元素對(duì)測(cè)定Pb 的干擾,降低了檢出限,但用于萃取的有機(jī)溶劑黏稠度較大,此類(lèi)溶劑用于上機(jī)測(cè)定干擾大,導(dǎo)致檢測(cè)結(jié)果穩(wěn)定性差,且有機(jī)溶劑會(huì)附著在設(shè)備進(jìn)樣系統(tǒng)內(nèi),難以清洗干凈,同時(shí)會(huì)腐蝕損壞設(shè)備,且通過(guò)燃燒揮發(fā)到空氣中,對(duì)操作人員、環(huán)境有一定的危害。且日方方法鉛、鎘的前處理方法和測(cè)定方法都不同,不能同時(shí)進(jìn)行檢測(cè),操作繁瑣,效率低。
也有一些報(bào)道使用以下幾種方法進(jìn)行測(cè)定:①通過(guò)在標(biāo)準(zhǔn)工作溶液中加入一定濃度的P 和Ca 基體,使用ICP-AES 光譜儀進(jìn)行同時(shí)測(cè)定[1];②通過(guò)固相萃取后,使用ICP-OES 光譜儀進(jìn)行同時(shí)測(cè)定[2];③X 射線熒光光譜標(biāo)準(zhǔn)添加法[3];④電感耦合等離子體質(zhì)譜法[4]。但第1種方法因每個(gè)樣品的基體組分、濃度不一,標(biāo)準(zhǔn)工作系列溶液中難以匹配至與樣品同樣組分和濃度的基體,基體匹配度不夠,產(chǎn)生的干擾不一致,對(duì)檢測(cè)結(jié)果準(zhǔn)確性產(chǎn)生一定的影響。第2種方法采用固相萃取法消除基體,但樣品溶液需要經(jīng)過(guò)萃取和洗脫過(guò)程,操作步驟繁瑣,耗時(shí)長(zhǎng)。第3種方法因X 射線熒光光譜法靈敏度較低,不適用于低含量成分的測(cè)定。第4種方法雖檢出限更低,但同樣需考慮基體匹配問(wèn)題,且ICP-MS 設(shè)備昂貴、操作維護(hù)要求高。
本文采用電感耦合等離子體發(fā)射光譜法-標(biāo)準(zhǔn)加入法同時(shí)測(cè)定磷礦中鉛鎘含量,通過(guò)標(biāo)準(zhǔn)加入法,達(dá)到基體匹配,消除了基體干擾,得到的檢出限較低,檢測(cè)結(jié)果準(zhǔn)確度較高,更適用于出口日本的磷礦檢測(cè),與日方檢測(cè)結(jié)果較為接近,并得到日方的充分信任和肯定。
1 實(shí)驗(yàn)部分
1.1 儀器及工作條件
電感耦合等離子體發(fā)射光譜儀。在儀器最佳工作條件下,凡能達(dá)到下列指標(biāo)者均可使用:
(1)光源:氬等離子體光源,發(fā)生器最大輸出功率不小于1.3kW。
(2)分辨率:200nm 左右時(shí)的光學(xué)分辨率優(yōu)于0.010nm:400nm 左右時(shí)的光學(xué)分辨率優(yōu)于0.020nm。
(3)儀器工作參數(shù)設(shè)定
高頻發(fā)生器功率為1.30kW;霧化氣流量為0.55L/min;輔助氣流量為0.20L/min;等離子體氣流量為15L/min;觀察高度為15mm;泵速為1.5mL/min;等離子體觀測(cè)方式為軸向觀測(cè);穩(wěn)定時(shí)間為30s;選擇自動(dòng)扣除各元素相應(yīng)背景點(diǎn)。
氬氣(體積分?jǐn)?shù)≥99.99%)。
1.2 試劑
鹽酸(ρ=1.19g/mL);硝酸(ρ=1.42g/mL);鉛標(biāo)準(zhǔn)貯存溶液(國(guó)家標(biāo)準(zhǔn)物質(zhì)中心):1 000μg/mL;鎘標(biāo)準(zhǔn)貯存溶液(國(guó)家標(biāo)準(zhǔn)物質(zhì)中心):1 000μg/mL;鉛、鎘混合標(biāo)準(zhǔn)溶液(10.0μg/mL):分別吸取鉛、鎘標(biāo)準(zhǔn)貯存溶液(1 000μg/mL)1.00mL于100mL 容量瓶中,加入5mL 硝酸(ρ=1.42g/mL),加水稀釋至刻度,混勻。
實(shí)驗(yàn)所用試劑為優(yōu)級(jí)純;實(shí)驗(yàn)用水為一級(jí)水。
1.3 分析步驟
1.3.1 試料的分解
稱取試樣5.0~10.0g(精確至0.01g)至于250mL 燒杯中,加少量水潤(rùn)濕,加入30mL 鹽酸,10mL 硝酸,加熱溶解可溶性鹽類(lèi),蒸發(fā)至溶液近干,取下燒杯,稍冷,加入5mL 硝酸、50mL 左右水,煮沸10min 左右,冷卻,轉(zhuǎn)移至100mL 容量瓶中,用水稀釋至刻度,混勻,干過(guò)濾,分別移取10mL 溶液于4個(gè)50mL 容量瓶中,再分別加入1.00、2.00、3.00mL 鉛鎘混合標(biāo)準(zhǔn)溶液(10.0μg/mL)、加入2mL 硝酸,用水稀釋至刻度,混勻,待測(cè)。隨同試料做空白試驗(yàn)。
1.3.2 測(cè)定
在電感耦合等離子體發(fā)射光譜儀上選定好波長(zhǎng),測(cè)定溶液中鉛、鎘的強(qiáng)度。以加入標(biāo)準(zhǔn)溶液濃度為橫坐標(biāo),對(duì)應(yīng)的強(qiáng)度為縱坐標(biāo),繪制標(biāo)準(zhǔn)曲線,用外推法(延長(zhǎng)標(biāo)準(zhǔn)曲線和橫坐標(biāo)相交的數(shù)的絕對(duì)值)就可以得到樣品液的濃度。
2 結(jié)果與討論
2.1 樣品處理方法的選擇
經(jīng)實(shí)驗(yàn)分析表明,單獨(dú)采用HNO3、HCl 均不能完全分解樣品,測(cè)定結(jié)果偏低;采用HF-HClO4,試樣能完全溶解,但HClO4沸點(diǎn)高,蒸發(fā)過(guò)程可能會(huì)造成鉛元素的損失,并且殘留的HClO4產(chǎn)生較強(qiáng)的背景干擾,影響測(cè)定結(jié)果[1];使用王水溶解樣品,溶解樣品效果更好,測(cè)定結(jié)果更準(zhǔn)確。
2.2 基體效應(yīng)和分析譜線的選擇
在磷礦中,成分元素多,基體非常復(fù)雜。特別是主量元素Ca、P,當(dāng)轉(zhuǎn)換成CaO、P2O5形式計(jì)算時(shí),其含量約為50%和35%左右,而待測(cè)元素Pb、Cd 的含量較低,因此基體干擾非常嚴(yán)重。且樣品溶液中的磷酸鹽具有黏稠性,欲通過(guò)增加稱樣量提高待測(cè)元素含量,可能會(huì)導(dǎo)致ICP 光譜儀的霧化器堵塞,嚴(yán)重影響結(jié)果的測(cè)定。本法通過(guò)標(biāo)準(zhǔn)加入法,達(dá)到基體匹配,消除基體干擾的作用。同時(shí)待測(cè)元素Pb 和 Cd 共存濃度均未達(dá)到造成干擾程度,故可同時(shí)測(cè)定。
選擇測(cè)定波長(zhǎng)時(shí)既要最大限度地避開(kāi)光譜干擾,又要考慮分析線的強(qiáng)度和準(zhǔn)確性。經(jīng)實(shí)驗(yàn)分析比較,選定鉛、鎘的測(cè)定波長(zhǎng)分別為220.3nm、228.8nm。這兩條分析譜線信背比高、干擾少、測(cè)定結(jié)果更準(zhǔn)確。
2.3 線性范圍和檢出限
在儀器最佳工作條件下,選擇標(biāo)準(zhǔn)加入法測(cè)定模式,對(duì)加入等量樣品的標(biāo)準(zhǔn)溶液系列進(jìn)行測(cè)定,以元素的質(zhì)量濃度為橫坐標(biāo),以待測(cè)元素凈發(fā)射強(qiáng)度為縱坐標(biāo),繪制校準(zhǔn)曲線。對(duì)樣品空白溶液進(jìn)行12次測(cè)定,以空白實(shí)驗(yàn)的3倍標(biāo)準(zhǔn)偏差所對(duì)應(yīng)的含量作為檢出限,10倍的檢出限為測(cè)定下限。各元素的線性范圍、線性相關(guān)系數(shù)、檢出限和測(cè)定下限結(jié)果詳見(jiàn)表1。
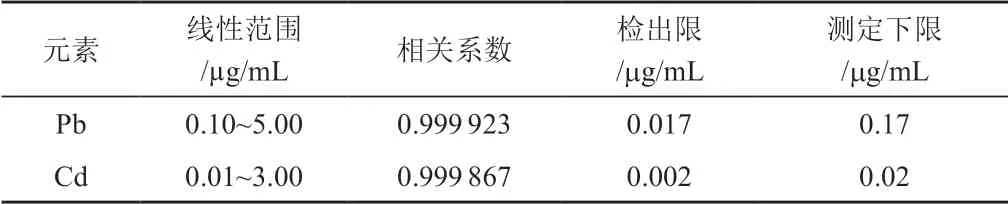
表1 校準(zhǔn)曲線的線性范圍、相關(guān)系數(shù)以及檢出限和測(cè)定下限
2.4 精密度和加標(biāo)回收試驗(yàn)
按照實(shí)驗(yàn)方法,分別對(duì)不同含量的3個(gè)典型磷礦樣品進(jìn)行了11次獨(dú)立測(cè)定,測(cè)定其鉛、鎘含量,計(jì)算其平均值及相對(duì)標(biāo)準(zhǔn)偏差,方法的相對(duì)標(biāo)準(zhǔn)偏差在1.87%~3.51%,精密度滿足要求。同時(shí)進(jìn)行加標(biāo)回收率試驗(yàn),回收率在96.6%~108.4%,滿足要求。
2.5 與日方結(jié)果比對(duì)
經(jīng)過(guò)對(duì)出口日本磷礦的鉛、鎘檢測(cè)數(shù)據(jù)進(jìn)行收集比對(duì),我方采用此法結(jié)果與日方結(jié)果均在檢測(cè)允差范圍內(nèi),具體比對(duì)數(shù)據(jù)見(jiàn)表2。

表2 比對(duì)試驗(yàn)
3 結(jié)語(yǔ)
用王水溶解樣品,硝酸浸提,將待測(cè)元素完全從樣品中溶解出來(lái)。采用標(biāo)準(zhǔn)加入法,在標(biāo)準(zhǔn)工作曲線系列溶液中加入等比的樣品量,使用電感耦合等離子體發(fā)射光譜儀進(jìn)行測(cè)定,以加入標(biāo)準(zhǔn)溶液濃度為橫坐標(biāo),對(duì)應(yīng)的強(qiáng)度為縱坐標(biāo),繪制標(biāo)準(zhǔn)曲線,用外推法(延長(zhǎng)標(biāo)準(zhǔn)曲線和橫坐標(biāo)相交的數(shù)的絕對(duì)值)得到樣品液的濃度。此法達(dá)到消除樣品基體干擾作用,方法操作簡(jiǎn)單,精密度好,準(zhǔn)確度高,特別適用于鉛、鎘含量較低的磷礦檢測(cè),完全滿足出口磷礦的檢測(cè)質(zhì)量要求,并彌補(bǔ)了國(guó)家標(biāo)準(zhǔn)在鉛、鎘低含量范圍測(cè)定方法的空缺。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
中學(xué)生數(shù)理化·七年級(jí)數(shù)學(xué)人教版(2021年6期)2021-11-22 07:50:58
當(dāng)代陜西(2019年8期)2019-05-09 02:22:48
動(dòng)漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
海峽科技與產(chǎn)業(yè)(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
專(zhuān)用汽車(chē)(2016年4期)2016-03-01 04:13:43
Coco薇(2015年1期)2015-08-13 02:47:34
