著色性干皮病兩例ERCC2和ERCC5基因突變分析
2020-06-05 06:53:52楊舟徐哲焦磊王珊馬琳
中華皮膚科雜志 2020年4期
關鍵詞:基因突變
楊舟 徐哲 焦磊 王珊 馬琳
國家兒童醫學中心 首都醫科大學附屬北京兒童醫院皮膚科100045
通信作者:馬琳,Email:bch_maleen@aliyun.com
著色性干皮病(xeroderma pigmentosum,XP)是一組罕見的常染色體隱性遺傳病,其發病機制為部分基因突變導致DNA 損傷修復能力受損,從而引發光敏、皮膚損傷、眼部病變、神經損害、惡性腫瘤、發育障礙等。皮膚癌為XP 患者的主要死因,神經退行性疾病是第二大死因[1]。目前報道XP 共有7個互補型(XPA、XPB、XPC、XPD、XPE、XPF、XPG)和1 個變異型(XPV),分別對應8 種基因(XPA、ERCC3、XPC、ERCC2、DDB2、ERCC4、ERCC5、POLH)突變[2]。本研究采用高通量測序對2 例XP患者進行全外顯子組測序(whole ? exome sequencing,WES),結合Sanger測序驗證,明確其分別為XPD型及XPG型。
資料與方法
一、病歷資料
例1 男,3 歲,出生時皮膚外觀正常,日曬后面部出現紅斑和腫脹,隨后皮膚粗糙、干燥,逐漸出現散在的雀斑樣病變和色素痣。皮疹夏季加重,以光暴露部位為主。近1年出現步態不穩,行走時易跌倒。體檢:面、耳、頸部和雙手背可見大量褐色斑點、斑片,間雜色素減退斑,散在疣狀丘疹,軀干散發黑褐色斑丘疹(圖1)。神經系統檢查:步態不穩,易跌倒,四肢肌張力正常,深部腱反射可引出;指鼻試驗、閉目難立征、輪替試驗均陰性,未見共濟失調;語言發育落后,無法表達完整的句子,智力篩查落后于正常同齡兒范圍。其余系統檢查未見明顯異常。眼科檢查正常;純音測聽未發現感音性耳聾;頭顱磁共振成像檢查大致正常,未見腦萎縮、腦室擴張、基底節及大腦皮質鈣化等。
例2 男,1 歲5 個月,生后3 個月日曬后面部出現輕度潮紅腫脹,后逐漸出現面部褐色斑點。體檢:面部散在褐色斑點、斑片,伴少許色素減退性斑(圖2)。體檢:神經及其他系統未見異常。眼科、聽力檢查正常。
2例患兒均為第1胎第1產,父母非近親結婚,無類似疾病家族史。囑患者防曬、避光,隨訪2年,2例患兒皮損均較前有所增多,未出現惡性腫瘤。
二、方法
1.標本采集:用乙二胺四乙酸二鉀抗凝管收集患兒及其父母外周靜脈血3 ml,使用北京天根生化科技有限公司DNA 提取試劑盒提取DNA(總量>10 μg,50 mg/L)。本研究符合2013年修訂的《赫爾辛基宣言》(https://www.wma.net/policies?post/wma?declaration?of?helsinki?ethical?principles?for?medical?research?involving?human?subjects/)的要求,患兒監護人及健康對照同意基因檢測并簽署知情同意書。
2.基因突變檢測:①WES測序,稀釋DNA后采用Covaris 超聲儀隨機分解成小片段DNA(180 ~280 bp),后經探針雜交和外顯子區序列捕獲,制備文庫并質檢,在北京康旭醫學檢驗所應用美國Illumina 公司的Hiseq X Ten 測序平臺進行測序,應用BWA(v0.7.15)軟件將測序read與人類參考基因組對比,應用GATK(v3.6)軟件檢測單核苷酸變異和插入/缺失突變;②注釋分析,使用ANNOVAR 注釋軟件注釋變異在基因和轉錄本中的位置,基因相關注釋涉及RefSeq、Ensembl、UCSC等數據庫,利用千人基因組、GnomAD、TOPMED 等數據庫注釋變異在人群中的頻率,使用蛋白損傷分析工具PolyPhen?2、SIFT 及Mutation Taster 進行致病性分析,利用OMIM、HGMD、ClinVar 等數據庫對疾病進行注釋,重點關注著色性干皮病相關基因XPA、ERCC3、XPC、ERCC2、DDB2、ERCC4、ERCC5 及POLH;③Sanger測序驗證,針對單基因所需要驗證的位點所在外顯子/內含子的上下游序列設計引物(表1),對患兒突變位點進行PCR擴增,反應條件:95 ℃預變性10 min,95 ℃變性30 s、60 ℃復性30 s、72 ℃延伸45 s、共35個循環,72 ℃延伸5 min。PCR產物純化后用ABI 3 730測序儀進行Sanger雙向測序,獲得數據后對結果進行判讀,與Ensembl genome browse 90公布的序列比對分析。進一步對患兒父母進行驗證,確定突變來源。
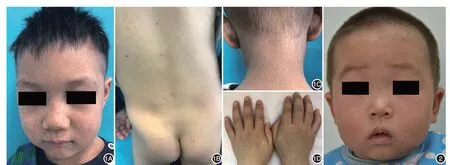
圖1 例1臨床表現 面部(1A)、背部(1B)、頸部(1C)、雙手背(1D)散發大量褐色斑點、斑片,伴色素減退斑,散發疣狀丘疹 圖2 例2面部散發褐色斑點、斑片,伴少許色素減退斑
結 果
例1存在ERCC2基因復合雜合突變,即父源突變c.1805G>A(p.Gly602Asp)和母源突變c.586C>T(p.Arg196Ter)(圖3),患兒符合XPD 型。例2 存在ERCC5 基因復合雜合突變,即父源突變c.2533+2T>C 和母源突變c.2453C>T(p.Ala818Val)(圖4),患兒符合XPG 型。例1 的ERCC2 基因突變c.586C>T導致編碼第196號氨基酸Arg的密碼子變成終止密碼子,為無義突變;例2 的ERCC5 基因突變c.2533+2T>C為編碼區第2 533位核苷酸后11位內含子中第2位核苷酸由T變為C,為剪切變異,經fruitfly 網站(www.fruitfly.org/seq_tools)剪切位點預測工具預測,突變將導致原有的剪切位點散失,很可能無法正常剪切,導致蛋白功能出現異常。以上兩個突變既往未見報道,在千人基因組、GnomAD、TOPMED 等健康人群對照數據庫中未發現或頻率極低,不屬于多態性位點。而ERCC2 基因突變c.1805G>A 和ERCC5 基因突變c.2453C>T 為已報道的致病性變異,使用SIFT、Polyphen?2和MutationTaster 軟件對2 個錯義突變c.1805G>A 和c.2453C>T 進行功能預測,均分別為“有害”、“很可能損害”和“致病性”,顯示其可能致病。Mutation Taster 軟件對無義突變c.586C>T 預測為“致病性”,可產生截短蛋白,影響蛋白功能。PolyPhen?2軟件顯示分析c.1805G>A、c.2453C>T 及c.586C>T 突變所在位點均在不同物種間高度保守。

表1 ERCC2和ERCC5基因Sanger測序引物序列
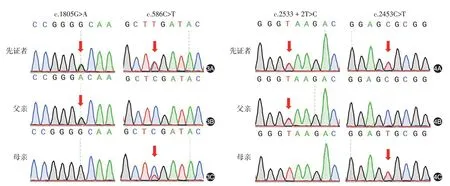
圖3 例1著色性干皮病患兒ERCC2基因復合雜合突變測序圖 3A:先證者發生錯義突變c.1805G>A(p.Gly602Asp)和無義突變c.586C>T(p.Arg196Ter);3B:先證者父親發生錯義突變c.1805G>A;3C:先證者母親發生c.586C>T無義突變 圖4 例2著色性干皮病患兒ERCC5基因復合雜合突變測序圖 4A:先證者發生(父源)剪切突變c.2533+2T>C和(母源)錯義突變c.2453C>T(p.Ala818Val);4B:先證者父親發生剪切突變c.2533+2T>C;4C:先證者母親發生錯義突變c.2453C>T
討 論
XPA ~XPG型XP是由涉及核苷酸切除修復機制的蛋白基因缺陷引起。NER可修復紫外線、化學試劑、毒性物質及化療藥物等引起的DNA損傷,以及一些因氧化還原過程產生的損傷[3]。不同分型的XP患者臨床表現有所差異,不同地域種族XP分型亦有差異。在日本XPA 占較大比例;在美國XPC 占優勢,其次為XPD 或XPV;在歐洲XPC 占主要比例,其次為XPV[4]。檢索HGMD Pro、PubMed及萬方和CNKI 數據庫顯示,截至2019 年10 月,我國XPA、XPC占優勢[4?7],XPV并不少見[5,7?8],而XPD僅報道2 例[5?6],XPG 報道5 例[5?7]。本文報道XPD、XPG各1例,其中XPD為我國少見類型。與其他地區人群XPG罕見相比,我國XPG并不少見,且本例患者癥狀較輕。
XPD(OMIM#278730)由ERCC2 基因突變引起5′?3′ATP 依 賴 的DNA 解 鏈 酶XPD 活 性 降 低 所致[9]。目前已報道100 余種與疾病相關的ERCC2基因突變,其基因型與表型關系復雜,可導致多種癥狀且嚴重程度差異很大,如XPD、XPD 合并Cockayne綜合征、毛發硫營養障礙、XP合并毛發硫營養障礙或腦-眼-面-骨骼綜合征[4,10]。XPD患者常早期發病,臨床表現嚴重(但輕于XPA),伴急性曬傷反應,雀斑樣色素沉著明顯,皮膚癌發生率較高[10]。XPD 患者多數可出現原發性神經元變性導致的進行性神經系統損害,如進行性步態不穩、肌張力低下、痙攣、癲癇等。這些癥狀開始于生命早期,亦可較晚出現,特征為髓鞘形成不足或異常、腦萎縮、鈣沉積和腦細胞異常[3]。患者神經系統癥狀不能完全用紫外線損傷修復缺陷解釋,還可能涉及核苷酸切除修復蛋白參與活性氧產生的損傷修復[3]。
我國既往僅報道2例XPD患者,均為復合雜合錯義突變所致,尚未發現神經系統癥狀,其中p.Gly47Arg 位于HD1 區,p.Ile595Ser、p.Gly602Asp、p.Arg666Trp 均位于HD2 區(圖5A)[5?6]。位于HD2區的突變可導致XP、XP 合并Cockayne 綜合征、毛發硫營養障礙或XP 合并毛發硫營養障礙表型[4]。本研究中例1 患兒存在錯義突變和無義突變的復合雜合突變,其錯義突變p.Gly602Asp 位于HD2 區域(圖5A),既往報道可引起XP合并Cockayne綜合征。另有研究者報道HD2 區中突變Arg683Trp 可導致XP表型及神經系統疾病進展[4],而Arg683Gln可導致神經系統輕度異常或晚期發作[10]。p.Arg196Ter 為無義突變,產生截短蛋白,且位于4FeS 簇區域(圖5A),推測其對酶活性影響較大。位于4FeS 簇區的突變可導致XP 或毛發硫營養障礙表型,其中I199Pfs*52 突變亦可導致XP 表型[11]。本文例1目前已出現大量雀斑樣色素沉著、皮膚異色、疣狀丘疹的XP皮膚表現,以及神經系統異常如步態不穩、語言及智力發育落后。有研究顯示,合并神經退行性疾病的XP 患者更早發生皮膚癌[2],故該患兒需嚴格避光防曬,預防皮膚腫瘤發生。XP 患者發生腦腫瘤的風險可增加50 倍,包括髓母細胞瘤、膠質母細胞瘤、脊髓星形細胞瘤和神經鞘瘤[3]。該患兒目前頭部磁共振成像檢查雖未見明顯異常,仍需密切觀察其神經系統癥狀進展。
XPG(OMIM 278780)是由ERCC5 基因突變引起結構特異性修復核酸內切酶XPG 活性缺陷所致,目前已報道50 余種ERCC5 基因突變與疾病相關。XPG 患者早期發病,伴急性曬傷反應,有明顯的雀斑樣色素沉著,可合并神經系統異常、Cockayne 綜合征或腦-眼-面-骨骼綜合征。其表型廣泛,可從輕微到極度受累和進行性神經元死亡導致的神經系統異常[12]。雖有報道本型患者于20歲可發生皮膚癌[1],但英國一項研究提示,XPG 患者皮膚癌的發生可能少于其他XP亞型[4]。
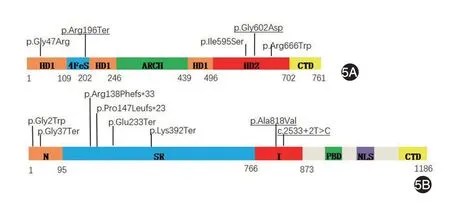
圖5 ERCC2和ERCC5蛋白結構及我國XPD和XPG型著色性干皮病患者基因突變位點 5A:ERCC2共5種突變;5B:ERCC5共8種突變。下劃線指示本研究中突變
我國既往報道的5 例XPG 患者中,3 例表現為輕度表型,暫未見腫瘤發生[5,7];1 例無義突變和錯義突變(p.Gln233Ter,p.Gly2Trp)患者合并恐懼癥、記憶力差及感覺神經性耳聾[13];1例復合雜合無義突變(p.Gln37Ter,p.Lys392Ter)患者合并智力減退[5]。本研究中例2 表現為輕度癥狀,存在剪切突變c.2533+2T>C 和錯義突變p.Ala818Val。盡管突變均位于核酸內切酶活性區域(I區)內(圖5B),但與無義或移碼突變相比,剪切突變和錯義突變對蛋白的影響可能相對較小,保留了一些殘留功能而導致輕癥。既往研究者推測ERCC5基因的蛋白截短突變可導致XPG/Cockayne 綜合征,攜帶至少1 個ERCC5 非截短突變的患者可能具有輕度XPG 表型[14],本研究結果支持該推測,并顯示I區的錯義和剪切突變亦可導致輕癥表型。但有文獻報道兩姐弟患者核酸內切酶I 區純合錯義突變p.Leu778Pro導致輕度XP 皮膚表型合并輕微神經異常[15]。本研究中患兒年齡較小,仍需進一步觀察是否出現遲發性神經系統癥狀。
本文提醒臨床醫生需提高對XP早期臨床表現的認識,以期早診斷、早防治。XPG 患者早期臨床表現可較為輕微;合并神經系統異常的患者除XPA外,應注意XPD 的可能。由于本病致病基因較多,臨床表型復雜,單獨應用Sanger 測序費時、費力且費用較高,采用高通量WES 結合Sanger 測序驗證的方法可快速、準確、經濟地進行基因診斷和分型,進一步指導預后及遺傳咨詢。
利益沖突 所有作者均聲明不存在利益沖突
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
