艾司氯胺酮與N-甲基-D-天冬氨酸受體的分子作用機制
2020-06-12 05:19:02張麗雷徐夢曉張靜曉張文曦唐文強
山東化工 2020年8期
關鍵詞:結構
張麗雷,徐夢曉,張靜曉*,張文曦,唐文強
(1.洛陽師范學院 化學化工學院,河南 洛陽 471934;2.洛陽師范學院食品與藥品學院,河南 洛陽 471934;3.陜西國際商貿學院醫藥學院,陜西 西安 712046)
抑郁癥通常表現為情緒低落,睡眠質量差,自尊心低下,以及失去對娛樂活動的興趣,并且具有高度復發的可能性。有研究表明重度抑郁癥的遺傳性為37%至48%[1],是嚴重影響人們正常生活的精神類疾病[2-4]。另外,抑郁癥是一種多因素疾病,主要與遺傳、生物學、環境和社會因素有關[5]。根據現代的心理學和生物學概念,抑郁癥不僅是精神障礙,而且也是生理疾病,具有明確的生物學基礎,例如重度抑郁癥病人會出現腦神經損傷,神經可塑性下降和神經元回路異常等生理問題[6],需要使用藥物進行治療。
氯胺酮是N-甲基-D-天冬氨酸受體(NMDAR)的拮抗劑,由于能夠快速改善抑郁癥狀和降低自殺意念,作為新型的抗抑郁藥物受到了廣泛的關注[7-9],但是由于具有致幻和濫用的風險限制了其臨床應用。艾司氯胺酮是氯胺酮的對映異構體,與NMDAR具有更強的結合力,并同樣具有改善抑郁癥狀和降低自殺意念的作用。然而,艾司氯胺酮抗抑郁的作用機制并不十分清楚,本文從艾司氯胺酮對NMDAR的拮抗機制出發,結合分子對接和分子動力學方法研究艾司氯胺酮與NMDAR的作用機制,為闡明艾司氯胺酮的抗抑郁機制和新型抗抑郁藥物的開發提供理論依據。
1 材料與方法
從蛋白晶體結構數據庫(RCSB PDB)中獲取NMDAR的晶體結構(3OEM),去除N-甲基-D-天冬氨酸配體和結晶水,利用PDB2PQR軟件包[10]補全缺失的原子,記為NMDAR,作為NMDAR的初始結構。從ATB數據庫[11]中獲取艾司氯胺酮的分子結構和力場數據(Molid:204037)。采用AutoDock Vina程序[12]對艾司氯胺酮和NMDAR進行對接,口袋大小設為20×20×20?3,網格間距設定為1.0?。
采用GROMACS(2018.4)軟件包[13]進行分子動力學模擬,將分子對接得到的艾司氯胺酮與NMDAR的復合物作為分子模擬的初始結構,采用GROMOS 54A7力場。在初始結構周圍延伸8?的立方體中填充水分子,建立周期性結構,并加入2個氯離子使體系保持中性,最終整個體系大約包含60000個原子。首先對整個體系進行1000步的能量極小化動力學,再進行100ps的限制性動力學,使溶劑弛豫,之后再進行20ns的NPT分子動力學模擬。在模擬過程中,設置時間步長2fs,運用Velocity-rescale方法控制整個體系的溫度為310K,采用parrinello-rahman方法控制體系的壓力為1bar,使用PME方法計算長程靜電相互作用,范德華截斷值設為12?。使用GROMACS軟件包對分子模擬的結果進行分析,并采用VMD軟件對結果進行可視化。
2 結果與討論
艾司氯胺酮與NMDAR蛋白對接的結果如圖1(a)所示,其中,艾司氯胺酮分子以球棍模型表示,NMDAR以蛋白的二級結構表示。從圖中可見,艾司氯胺酮分子成功對接于蛋白C端(CT)葉片和N端(NT)葉片中間的口袋位置。考察了艾司氯胺酮的結合引起的NMDAR結構和運動趨勢的變化,NMDAR對接前后模擬組合軌跡的主成分porcupine圖如圖1(b,c)所示,其中,箭頭表示在蛋白質在第一振動模式下骨架原子的運動方向。從圖中可見,對接之前的NMDAR蛋白的運動主要是C端和N端葉片結構在逆時針方向上的有序運動,其中NT的運動振幅更大,C端振幅較小,運動更加無序。當NMDAR與艾司氯胺酮對接之后,蛋白NT的運動改為順時針的運動,并且運動趨勢減弱,也更加無序。這可能是由于艾司氯胺酮的對接減少了有序剛體葉片的旋轉運動程度,起到了穩定蛋白結構的作用。

圖1 艾司氯胺酮與NMDAR對接后的結構(a),和NMDAR對接前(b)后(c)分子模擬運動的porcupine圖Fig.1 Docking configuration of s-ketamine with NMDAR,and porcupine diagrams of the NMDAR before (b) and after (c) docking
為了確定分子模擬過程中蛋白和配體的穩定性,考察了模擬過程中蛋白和配體結構的均方根偏差(RMSD),結果如圖2所示。從圖中可見,經過約3ns的模擬之后,NMDAR對接前后的結構中蛋白和配體的RMSD值均趨于穩定,上下波動范圍穩定在2?范圍之內,表明所研究結構在經過平衡后均達到穩定結構。NMDAR對接前后結構的RMSD沒有發生明顯的改變,表明艾司氯胺酮的對接對NMDAR的整體結構影響不大。另外,模擬過程中艾司氯胺酮的RMSD值變化較小,表明艾司氯胺酮的結構和位置沒有發生明顯的變化,結合穩定。

圖2 NMDAR與艾司氯胺酮對接前后分子模擬過程中的RMSD圖Fig.2 RMSD of NMDAR before and after docking withs-ketamineduring molecular simulation
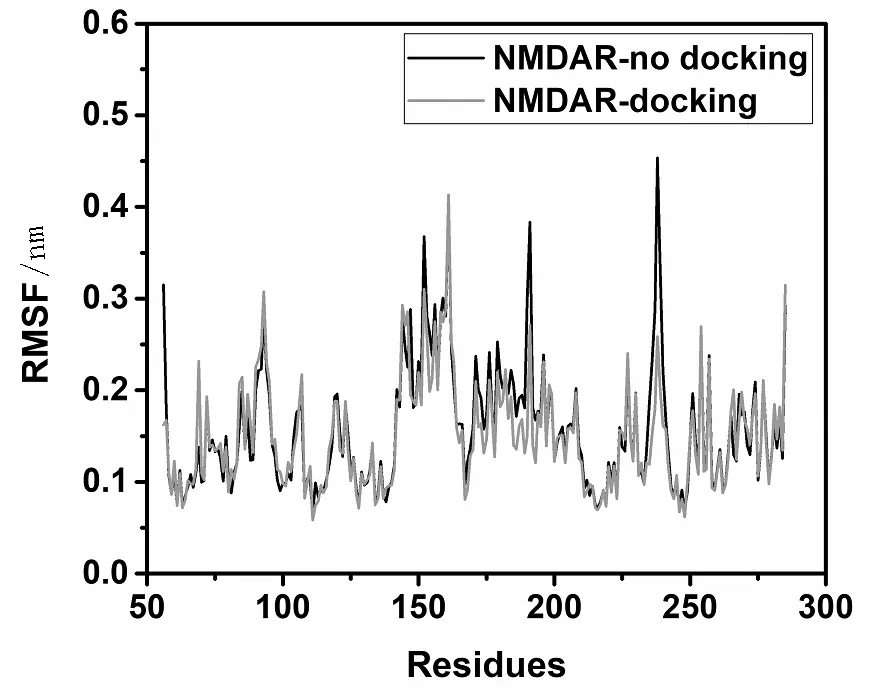
圖3 NMDAR與艾司氯胺酮對接前后結構分子模擬過程中的RMSF圖Fig.3 RMSF of NMDAR before and after docking with s-ketamine during molecular simulation
蛋白殘基的均方根波動值(RMSF)是衡量殘基在整個運動軌跡中相對于平均位置的波動程度。為了考察艾司氯胺酮的結合對NMDAR功能區域的影響,對NMDAR在結合前后分子模擬過程中各個殘基的RMSF進行了分析,結果如圖3所示。從圖中可見,在艾司氯胺酮的結合之后,部分殘基的RMSF值發生了明顯的變化。其中,TRP238、THR56、GLY190、LYS239、LYS191、ARG237等殘基的RMSF變化較大,表明艾司氯胺酮的結合可能主要影響與這些殘基相關區域的功能,從而調節NMDAR的活性。
通過考察蛋白的溶劑可及表面積可以考察蛋白的親水和疏水性。考察了NMDAR在與艾司氯胺酮對接前后的結構在分子模擬過程中溶劑可及表面積的變化,結果如圖4所示。從圖中可見,蛋白的溶劑可及表面積可分為親水性表面積和疏水性表面積,NMDAR在與艾司氯胺酮對接之后,總的溶劑可及表面積有所減小,其中,疏水性表面積幾乎無變化,而親水性表面積減小,表明NMDAR在與艾司氯胺酮對接之后,蛋白的親水性減小,這可能是艾司氯胺酮能夠抑制NMDAR活性的原因之一。

圖4 NMDAR與艾司氯胺酮對接前后分子模擬的溶劑可及表面積Fig.4 Solvent accessible surface of NMDAR before and after docking with s-ketamine during molecular simulation

圖5 NMDAR與艾司氯胺酮對接前后分子模擬過程中蛋白內部的氫鍵數目Fig.5 Hydrogen bonds number inside NMDAR before and after docking with s-ketamine during molecular simulation
蛋白內部的氫鍵是影響蛋白穩定性的主要原因之一。為了考察艾司氯胺酮對NMDAR穩定性影響的原因,分析了MBDAR在與艾司氯胺酮對接前后蛋白內部氫鍵數目的變化,結果如圖5所示。從圖中可見,在與艾司氯胺酮對接之前,MBDAR內部的氫鍵數目在190個左右,在分子模擬的過程中變化不大,維持在170個至210個之間。在與艾司氯胺酮對接之后,MBDAR內部的氫鍵數目有所增加,并且在模擬的過程較為穩定,維持在180個至215個之間,表明MBDAR在與艾司氯胺酮對接之后蛋白內部氫鍵數目的增加可能是蛋白穩定的原因。
3 結論
采用分子對接和分子動力學方法研究了艾司氯胺酮與NMDAR之間的分子作用機制,結果表明,艾司氯胺酮趨向結合于NMDAR的口袋部位,使蛋白N端的運動方式從有序的順時針運動改變為無序的運動,并使運動振幅減弱。經過分子模擬獲取了NMDAR與艾司氯胺酮結合的穩定結構,發現艾司氯胺酮主要影響蛋白的TRP238、THR56、GLY190等殘基部位。通過蛋白的溶劑可及表面積和蛋白內部氫鍵數目分析表明,艾司氯胺酮的結合使NMDAR的親水性減小,疏水性增加,蛋白內部氫鍵數目增加,這可能是艾司氯胺酮調節NMDAR結構活性的主要因素,本文的研究為闡明艾司氯胺酮的抗抑郁機制和新型抗抑郁藥物的開發提供了理論依據。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
