恩曲他濱的合成工藝優化
2020-06-27 08:20:16顧繼山吳曉宇孫國鋒丁一剛劉生鵬
武漢工程大學學報 2020年2期
李 蘇,顧繼山,吳曉宇,熊 蕓,孫國鋒,丁一剛,劉生鵬
武漢工程大學化工與制藥學院,湖北 武漢 430205
恩曲他濱(emtricitabine,FTC),化學名為4-氨基-5-氟-1-[(2R,5S)-2-羥甲基-1,3-氧硫雜環戊烷-5-基]-2(1H)-嘧啶酮,是一種核苷類逆轉錄酶抑制劑。該藥由美國Gilead 公司開發,于2003 年經美國FDA 批準上市,用于治療由人類免疫缺陷病毒(HIV)感染引起的艾滋病,以及乙型肝炎病毒(HBV)引起的乙型肝炎[1]。自上市以來,臨床證明FTC 的抗病毒失敗率低,治療效果好,是治療艾滋病和乙型肝炎最成功的藥物之一[2-3]。
傳統的FTC 濱合成路線主要采用化學合成的方法[4-6],總收率均在29.8%~51%之間,且都存在成本高、反應步驟多、毒性大和污染環境等缺點[7-8]。目前,由于脂肪酶具有高度的立體選擇性、熱穩定性、底物廣泛性、催化活性、環境友好性和回收利用性等[9-11],而被廣泛用于藥物的合成中[12-14]。本文在參考已報道的工藝基礎上[8,12,15],以苯甲酸鈉2 和溴乙醛縮二乙醇3 為原料,縮合得到苯甲酰氧基乙醛縮二乙醇4,將4 在濃度為2.4 mol/L 鹽酸溶液中水解得到苯甲酰氧基乙醛5,5 與1,4-二噻烷-2,5-二醇6 在脂肪酶Novozyme 435 催化下得到(2R,5S)-2-苯甲酰氧甲基-5-乙酰氧基-l,3-氧硫雜環戊烷7,7 與硅烷化5-氟胞嘧啶8 進行偶聯得到4-氨基-5-氟-1-[(2R,5S)-2-苯甲酰氧甲基-1,3-氧硫雜環戊烷-5-基]-2(1H)-嘧啶酮9,將其水解后可得終產物FTC(圖1),總收率為51.3%,FTC 純度為99.7%。
1 實驗部分
1.1 儀器與試劑
Bruker Avance NEO-600 MHz 型核磁共振波譜儀(德國Bruker 科技有限公司);SHIMADZU CT0-20AC 高效液相色譜儀,SHIMADZU-2014 氣相色譜儀(日本島津儀器公司);WRS-3A 熔點儀(上海儀電物理光學儀器有限公司);JASCO P-1030 自動旋光儀(日本分光株式會社)。
苯甲酸鈉,1,4-二噻烷-2,5-二醇,5-氟胞嘧啶(阿拉丁生化科技股份有限公司);溴乙醛縮二乙醇,三乙烯二胺,六甲基二硅胺烷(武漢格奧化學技術有限公司);鹽酸,乙酸苯酯,正己烷,無水乙醇,三氟乙酸(上海麥克林生化科技有限公司);四氫呋喃,N,N-二甲基甲酰胺,乙酸乙酯,二氯甲烷,三乙胺,甲醇等,(國藥集團化學試劑有限公司);所有試劑均為分析純。Novozyme 435(諾維信公司)。
1.2 結果檢測
色譜柱:Chiralpak AD-H 柱(250 mm×4.6 nm,5 μm);流動相:V(正己烷)∶V(無水乙醇)∶V(甲醇)∶V(三氟乙酸)∶V(三乙胺)=800∶150∶50∶1∶1;柱溫:40 ℃;檢測器波長280 nm;流速設定為1.0 mL/min,進樣量20 μL。精密稱取合成的FTC約5.0 mg,加入1 mL 甲醇溶解,轉移至10 mL 容量瓶中,并用流動相稀釋至刻度,制成濃度約為0.5 mg/mL 的溶液。結果得出目標產物恩曲他濱1的純度為99.7%,符合中國藥典標準。
1.3 實驗方法
1.3.1 苯甲酰氧基乙醛縮二乙醇(4)的合成 稱取化合物2(9.925 g,68 mmol)于1 L 三口燒瓶中,并加入N,N-二甲基甲酰胺(N,N-Dimethyl formamide,DMF,300 mL),加入3(19 mL,124.9 mmol),攪拌溶解,于135 ℃回流反應2 h 后,再加入化合物2(9.925 g,68 mmol)繼續反應14 h,TLC 監測3消失。反應完畢后,旋蒸除去DMF,加入乙酸乙酯溶解,分別用蒸餾水和飽和氯化鈉溶液洗滌,萃取有機層,無水硫酸鈉干燥,旋蒸除去溶劑,得到紅色粘稠液體26.25 g,收率88.2%。1H NMR(600 MHz,DMSO-d6),δ:8.07(d,2H,J=8.0 Hz,Ar-H),7.58(t,1H,J=7.4 Hz,Ar-H),7.45(t,2H,J=7.7 Hz,Ar-H),4.85(t,1H,J=5.5 Hz,-CH),4.36(d,2H,J=5.5 Hz,-CH2),3.71(q,4H,-CH2),1.23(t,6H,J=7.1 Hz,-CH3)。13C NMR(600 MHz,DMSO-d6),δ:166.27,133.06,130.04,129.73,128.39,99.75,64.47,62.54,15.36。與文 獻[8]一致。

圖1 FTC 的合成路線Fig.1 Synthesis route of FTC
1.3.2 苯甲酰氧基乙醛(5)的合成 稱取化合物4(3.0 g,12.6 mmol)、20 mL 四氫呋喃于50 mL 三口燒瓶中,升溫至60 ℃回流反應,磁力攪拌下,滴加入2.4 mol/L(10 mL)的鹽酸。1 h 后,結束反應。旋蒸除去四氫呋喃,加入乙酸乙酯溶解,用飽和碳酸氫鈉溶液和氯化鈉溶液洗滌,萃取有機層,無水硫酸鈉干燥,旋蒸除去溶劑。粗品經層析柱純化,得到淺黃色油狀液體1.77 g,收率為84.7%。1H NMR(600 MHz,DMSO-d6),δ:9.73(s,1H,-CHO),7.96~8.17(d,2H,J=7.45 Hz,Ar-H),7.61(d,1H,J=7.2 Hz,Ar-H),7.48(d,2H,J=7.7 Hz,Ar-H),4.91(s,2H,-CH2)。13C NMR(600 MHz,DMSO-d6),δ:196.11,166.21,133.86,130.15,129.14,128.78,69.26。與文獻[8]一致。
1.3.3 (2R,5S)-2-苯甲酰氧甲基-5-乙酰氧基-1,3-氧硫雜環戊烷(7)的合成 稱取化合物5(0.26 g,1.58 mmol)、6(0.14 g,0.95 mmol)、乙酸苯酯(0.6 mL,4.74 mmol)、三乙胺(0.22 mL,1.43 mmol)、DABCO(0.013 g,0.094 8 mmol,0.1 eq)溶于10 mL 無水四氫呋喃中,40 ℃回流反應6 h。向反應混合物中加入0.38 g 0.4 nm 分子篩,緩慢攪拌1 h,加入磷酸鹽緩沖溶液(pH=7.4,3 mL),250 mg(25 mg/mL)Novozyme 435 繼續攪拌8 h 后停止。過濾反應混合物,濾液用飽和氯化鈉溶液洗滌,萃取有機層,無水硫酸鈉干燥,旋蒸除去溶劑。粗品經層析柱純化,得到無色油狀液體0.371 g,收率為82.1%,ee值為99%(文獻[8]為82%)。1H NMR(600 MHz,DMSO-d6),δ:8.05(d,2H,J=8.0 Hz,Ar-H),7.55(d,1H,J=7.2 Hz,Ar-H),7.43(d,2H,J=7.5 Hz,Ar-H),5.43(t,1H,J=4.7 Hz,-CH),4.75~4.94(m,1H,-CH),4.01~4.57(m,2H,-CH2),2.97~3.24(m,2H,-CH2),2.04(s,3H,-CH3)。13C NMR(600 MHz,CDCl3),δ:171.31,166.01,133.21,129.75,128.44,100.06,81.10,66.42,38.69。與文獻[12]一致。
1.3.4 4-氨基-5-氟-1-[(2R,5S)-2-苯甲酰氧甲基-1,3-氧硫雜環戊烷-5-基]-2(1H)-嘧啶酮(9)的制備 稱取5-氟胞嘧啶(0.52 g,4.0 mmol)、3 mL 六甲基二硅胺烷、0.1 g 催化劑硫酸銨、15 mL DMF 于100 mL 三口燒瓶中。升溫至100 ℃,回流反應。反應至反應液澄清透明,停止反應并旋蒸除去六甲基二硅胺烷,冷卻至室溫,得到化合物8。加入1.4 mL 三乙胺混合均勻以備用。
將化合物7(0.3 g,1.0 mmol)溶于6 mL 無水二氯甲烷中,倒入上述制備的8,并加入0.3 g 孔徑0.4 nm 分子篩,于80 ℃下回流攪拌反應8 h。反應結束后,加入乙酸乙酯溶解,用飽和碳酸氫鈉溶液、蒸餾水和鹽水洗滌,萃取有機層,無水硫酸鈉干燥,旋蒸除去溶劑,用乙醇重結晶,得到粗產物混合物,為淺黃色粉末。不經純化,直接進入下一步。1.3.5 FTC(1)的合成 取化合物9(0.3 g,0.7 mmol)溶于50 mL 甲醇和50 mL 氨水溶液中混合,在室溫下攪拌10 h 后,抽濾,旋蒸除去溶劑,粗品經層析柱純化,得到0.15 g 白色粉末,收率為83.6%,純度為99.7%,ee 值為99.5%。旋光度[α]25D=-119.4°(c 1.05,MeOH)[文 獻[4]為-116°,c 1.05,MeOH];m.p.180~185 ℃;1H NMR(600 MHz,DMSO-d6),δ:8.19(d,1H,J=7.2 Hz,-CH),7.81(s,1H,NH),7.57(s,1H,NH),6.13(s,1H,NH),5.41(t,J=5.54 Hz,1H,-OH),5.17(t,1H,J=3.8 Hz,-CH),3.65~3.82(m,2H,-CH2),3.39(dd,1H,J=24.5 Hz,-CH2),3.12(dd,1H,J=11.9 Hz,4.3 Hz,-CH2)。13C NMR(600 MHz,DMSO-d6),δ:157.95,153.45,137.06,135.47,126.26,87.01,62.60,37.24。與文獻[7]一致。
2 結果與討論
2.1 化合物5 反應條件優化
考察了以88%甲酸水溶液、不同濃度鹽酸和對甲苯磺酸等作為H+供體時的反應條件(表1)。可以得出,當反應溫度由室溫升至60 ℃時,副產物乙醇蒸發,使反應向正反應方向進行。并且,采用2.4 mol/L 鹽酸時,得到最高收率,88.2%。

表1 用于縮醛水解反應的不同催化劑的反應條件和結果*Tab.1 Reaction conditions and results over different catalysts used for hydrolysis reaction
2.2 脂肪酶催化合成化合物7 的優化
2.2.1 溶劑對酶催化反應的影響 酶促催化反應中,酶的催化活性和立體選擇性會受到溶劑的logP 值和極性的影響,因此必須選擇合適的有機溶劑。選取除水處理后的甲苯、二氯甲烷、四氫呋喃、乙腈、甲醇和N,N-二甲基甲酰胺6 種有機溶劑進行對比。從表2 可以看出,四氫呋喃作為溶劑時,轉化率和對映體選擇性最高。在強親水性溶劑(logP <0)中,反應的轉化率很低,是因為親水性溶劑導致Novozyme 435 脂肪酶周圍必需的水層被剝離,脂肪酶的結構被破壞,催化活性降低。而非極性溶劑(logP >2.5)的疏水性雖然能使酶結構穩定,但是其對映體選擇性很低。
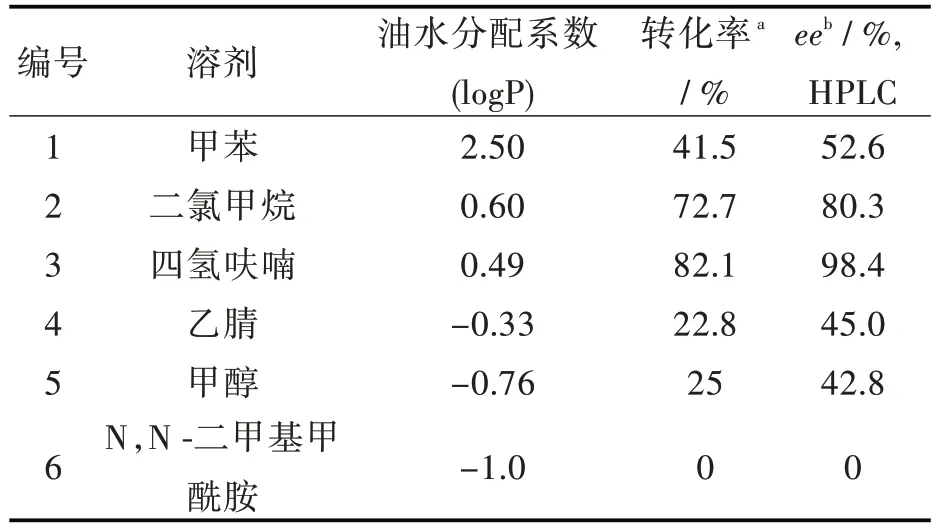
表2 溶劑對酶催化反應的影響Tab.2 Effect of solvent on reaction of lipase-catalysis
2.2.2 溫度對催化反應的影響 由于脂肪酶的本質是蛋白質,溫度會影響酶的活性、穩定性和對映體選擇性,所以必須考察溫度對脂肪酶的催化反應的影響。通常情況下,隨著溫度升高,反應速度也隨之升高。當升高到一定溫度后,酶逐漸變性,失去催化活性。本研究考察了25~70 ℃之間的影響,結果如表3 所示。可以看到,轉化率在40 ℃時達到最大值,因此選取40 ℃為最佳反應溫度。

表3 溫度對酶催化反應的影響Tab.3 Effect of temperature on lipase-catalysis reaction
3 結 論
本文優化了FTC 的合成工藝路線。優化了苯甲酰氧基乙醛5 的合成工藝;以及在關鍵中間體7的合成過程中,選取Novozyme 435 脂肪酶為催化脂肪酶用于催化反應;在催化過程中確定了最佳反應溶劑和反應溫度,分別為四氫呋喃和40 ℃,得到ee 值>99%,收率為82.1%的化合物7。經過優化后,得到總收率為51.3%,純度為99.7%的FTC。綜上所述,本文的研究內容為其他不對稱手性化合物的合成提供了一種有參考價值的思路和方案。
