無分散劑微波輔助離子液體分散液液微萃取/膠束電動色譜檢測食用油中丙烯酰胺和5-羥甲基糠醛殘留
2020-07-03 03:41:16高仕謙吳友誼毛宇成
分析測試學報 2020年6期
關鍵詞:方法
徐 蕊,高仕謙,吳友誼*,毛宇成
(1.蘇州科技大學 環境科學與工程學院,江蘇 蘇州 215009;2.江蘇省環境科學與工程重點實驗室,江蘇 蘇州 215009)
含有糖和蛋白質的食品在高溫(>120 ℃)加熱期間(例如烘烤、油炸、燒烤等)會發生美拉德反應(褐變反應)[1],其中間產物丙烯酰胺(Acrylamide,AA)和5-羥甲基糠醛(5-Hydroxymethylfurfural,5-HMF)由于在食品中廣泛存在且毒理效應顯著而倍受關注。據報道[2],AA具有神經、肝和基因毒性作用;5-HMF對人類和動物有染色體致突變性和致癌性。我國食用植物油生產多以含有大量蛋白質和糖類(或稱碳水化合物)的油籽為原料。AA和呋喃類有害物質會在高溫焙炒(150~180 ℃)油籽的過程中產生[3-4],但目前有關AA和5-HMF的分析研究中,樣品基質集中于谷物類(如麥片、面包、薄脆餅干)、奶粉、牛奶、咖啡和馬鈴薯類等產品,食用油基質中此兩種目標物的研究非常缺乏。且現有植物油中對5-HMF進行分析研究[5-7]的實驗均需使用大量正己烷等有機溶劑,不環保且操作復雜。已有專利[8]利用油脂受熱誘導所產生的物質如丙烯酰胺、糠醛、呋喃類等的含量區分商品植物油、地溝油、煎炸老油,但尚未發現食用油中AA的相關檢測研究。因此建立食用油中AA和5-HMF的分析檢測方法對食用油質量管控、食用油安全和營養健康等具有非常重要的實際意義。
目前常用的檢測AA和5-HMF的方法包括高效液相色譜法、液相色譜-質譜法、氣相色譜法、氣相色譜-質譜法,但操作均較為復雜、檢測成本高,并且難以對復雜樣品基質中的微痕量目標物進行快速準確檢測[9]。近年來,毛細管電泳(Capillary electrophoresis,CE)因具有分析速度快、分離效率高、環境友好(試劑消耗微量)和檢測成本低等優勢,被認為是食品科學領域高效的分析工具[10]。食用油基質復雜,需要前處理以富集和凈化。常用的AA和5-HMF的前處理技術,如固相萃取[11]、液液萃取[12]、液液微萃取[7]等方法或多或少會使用有機溶劑且耗時,因而開發簡捷且環保的方法極為必要。分散液液微萃取(Dispersive liquid-liquid microextraction,DLLME)相比于傳統方法具有簡單、快速、成本低、回收率高等優點,自2006年被提出后倍受關注[13-14],且已成功應用于各種基質中痕量污染物的分析。然而,DLLME的萃取劑和分散劑大多采用有機溶劑,限制了其發展應用[15]。而離子液體(Ionic liquid,IL)由于低揮發性和低毒性被越來越多地用作DLLME的萃取溶劑[16]。IL-DLLME融合了以上兩者的優點,而且IL可有效吸收微波能量,采用微波輔助(Microwave assistant,MA)將更有利于IL萃取分析物[17]。另外,該方法無需分散劑,只需借助微波儀自帶的磁力攪拌作用即可達到較好分散效果,且開發免分散劑的DLLME方法也將是未來發展的一種趨勢[16,18]。
本研究將結合IL-DLLME、MA以及CE的優勢,建立一種基于離子液體和免分散劑的微波輔助萃取結合毛細管膠束電動色譜(MEKC)同時分析檢測食用油中AA和5-HMF的簡捷、綠色的方法。
1 實驗部分
1.1 材料與儀器
AA標準品(純度>99.8%,阿拉丁公司);5-HMF標準品(純度>98%,阿法埃莎公司);1-乙基-3-甲基咪唑四氟硼酸鹽([C2MIM][BF4])、溴化 1-丁基-3-甲基咪唑([C4MIM]Br)、溴化1-己基-3-甲基咪唑([C6MIM]Br)、溴化1-辛基-3-甲基咪唑([C8MIM]Br)(純度均為99%,上海成捷化學有限公司);甲醇(色譜純,美國Sigma公司);氫氧化鈉、鹽酸、十二烷基硫酸鈉(SDS)、四硼酸鈉(分析純,南京化學試劑股份有限公司)。
P/ACETMMDQ毛細管電泳儀配PDA檢測器(美國貝克曼公司);UWave-2000 多功能微波合成萃取儀(上海新儀微波化學科技有限公司);KQ-300E 型超聲波清洗器(昆山市超聲儀器公司);SK-1快速混勻器(金壇市科析儀器有限公司);TGL-16C 臺式離心機(上海安亭科學儀器廠);AL204 電子分析天平(梅特勒-托利多儀器上海有限公司);高溫鼓風干燥箱(上海姚氏儀器設備廠);PHS-3C臺式pH計(上海儀電科學儀器股份有限公司);熔融石英毛細管(河北銳灃色譜器件有限公司)。
1.2 實驗方法
1.2.1 標準溶液的配制分別準確稱取25 mg AA和5-HMF標準品于25 mL棕色容量瓶中,用甲醇定容后得到1 000 μg/mL的混標儲備液,密封儲存于-4 ℃下待用。各濃度混標使用液由儲備液加甲醇稀釋得到。
1.2.2 樣品采集芝麻油、金龍魚大豆油(精煉一級)和菜籽油購自校園周邊超市,其中大豆油不含AA和5-HMF,作為樣品空白。花椒油來自校園周邊餐館自制,煎炸老油來自校園周邊炸串小吃攤,廢餐廚油粗制品為蘇州某餐廚油脂回收公司回收的廢棄餐廚油經分選、濕熱分解后得到的油。
1.2.3 樣品前處理在2 mL油中,加180 μL的[C8MIM]Br離子液體和一個小磁子,于70 ℃下微波輔助分散液液微萃取12 min,置于-4 ℃冰箱中冷凍5 min,用磁體沿管壁緩慢取出轉子,以10 000 r/min高速離心5 min。定量移取160 μL下相后,加硼砂溶液(20 mmol/L,pH 9.7)使溶液總體積為400 μL,渦旋混勻1 min,10 000 r/min高速離心5 min后取下相,經0.22 μm針式PVDF濾頭(天津市津騰試驗設備有限公司)過濾,進CE分析。加標樣品的處理與上述步驟相似,但需先在油樣中加入一定量的混標。
1.2.4 膠束電動色譜條件實驗使用未涂覆的熔融石英毛細管(內徑50 μm,總長度49.2 cm,有效長度38.5 cm)進行電泳分離。所有溶液均經0.22 μm濾頭過濾。每天進樣前,依次用1 mol/L NaOH、水、運行緩沖液(BGE)沖洗毛細管 8、5、6 min;兩次運行間用BGE沖洗2 min。分離電壓為12 kV;0.5 psi(3 447.39 Pa)壓力下進樣6 s;檢測波長為202 nm;溫度25 ℃;緩沖溶液:含130 mmol/L SDS的20 mmol/L硼砂溶液(pH 9.7)。
2 結果與討論
2.1 膠束電動色譜條件的優化
毛細管電泳條件的優化因子包括緩沖液pH值和濃度、十二烷基硫酸鈉(SDS)的濃度、分離電壓、進樣時間和毛細管柱溫。
2.1.1 緩沖液pH值及濃度考察了硼砂和磷酸鹽緩沖液對目標物分離效果的影響,發現磷酸鹽的電流相對較大且目標物響應和分離效果欠佳。因此,選擇硼砂緩沖液并考察了其pH值的影響,結果顯示,在pH 9.2~10.8范圍內,隨著pH值的增加,目標物遷移時間延長,峰形變寬;當硼砂緩沖液為pH 9.7 時,兩目標物的響應和分離效果最好,因此選擇pH 9.7進行后續研究。在0~20 mmol/L范圍內,硼砂溶液濃度越大,目標物遷移時間越長,目標物峰面積越大,當硼砂濃度大于20 mmol/L后,電導率增加,基線不穩,致使目標物響應重現性變差,因而采用 20 mmol/L硼砂溶液作為緩沖溶液。
2.1.2 SDS濃度在分離過程中,由SDS形成的膠束可通過與目標物之間的疏水和靜電相互作用改善目標物的響應和分離情況。本文考察了緩沖液中不同濃度(10~150 mmol/L)的SDS對目標物分離的影響。發現高濃度的SDS可改善目標物的分離情況,但也導致焦耳熱增加,使得信噪比和重現性變差,最終選擇含有130 mmol/L SDS的20 mmol/L硼砂溶液(pH 9.7)作為最佳緩沖液。
2.1.3 分離電壓考察了分離電壓在10~20 kV范圍內對各組分遷移時間的影響。結果發現,隨著電壓的增加,遷移時間因電滲流(EOF)的增加而減小。然而,由于較高電壓下的焦耳熱顯著,導致目標物響應不佳以及基線漂移。綜合考慮后選擇12 kV作為分離電壓。
2.1.4 進樣時間在進樣壓力為0.5 psi(3 447.39 Pa)條件下,考察了進樣時間在2~10 s范圍內的影響。當進樣時間為6 s時,得到分析物最佳的峰面積和重現性,當進樣時間繼續增加時,僅觀察到峰高略有增加,且峰寬變寬,峰形尖銳性變差。因此最終選擇6 s為進樣時間。
2.1.5 毛細管柱溫考察了毛細管柱溫(15、20、25、30 ℃)對分離效果的影響,發現柱溫對目標物的分離效果影響相對較小。隨著柱溫升高,溶液粘度降低,出峰時間縮短,電滲流加快,影響準確性和重現性,因此選擇25 ℃為最佳柱溫。
2.2 萃取條件的優化
為了獲得最佳的萃取效果,通過單因子優化考察評估了一系列變量,包括離子液體的種類和體積、微波輔助萃取條件和樣品油體積,所有實驗均重復3次。
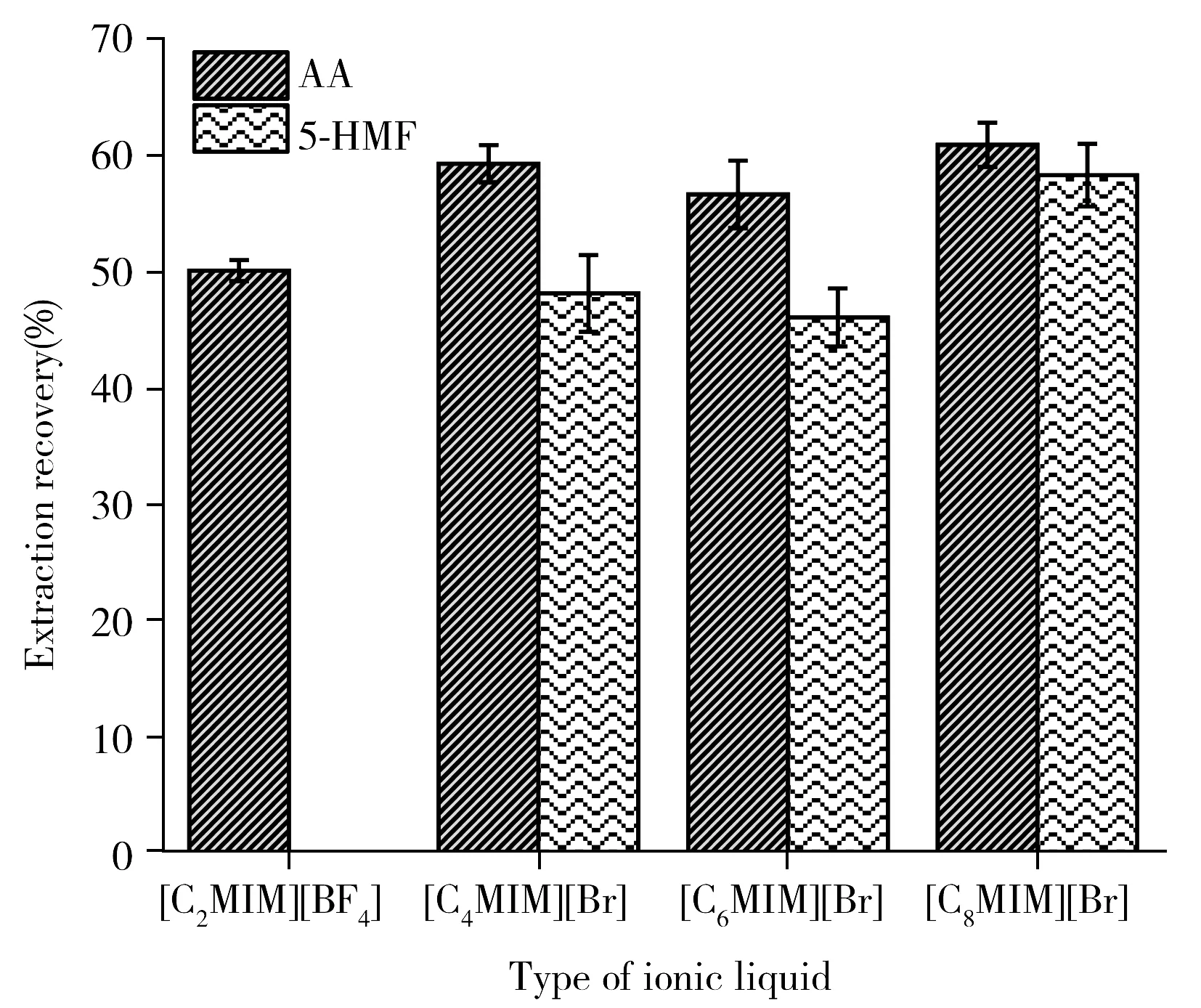
圖1 離子液體種類對萃取效果的影響Fig.1 Effect of ionic liquid types on extraction recovery
2.2.1 離子液體種類IL通常由相對較大的有機陽離子和單個或復雜的陰離子組成。IL的結構對其理化性質有重要影響,例如密度、粘度、在樣品溶液中的溶解度等,這些性質會強烈影響目標分析物的提取效率。本研究對4種咪唑類IL,包括[C2MIM][BF4]、[C4MIM]Br、[C6MIM]Br和[C8MIM]Br進行了考察,發現[C2MIM][BF4]對5-HMF無提取能力。其它3種IL具有相同陰離子但陽離子(主要是烷基鏈長度)不同。有研究表明,離子液體的疏水性主要取決于其陽離子的性質,即烷基鏈長度越長疏水性越好[18]。而本實驗結果(圖1)表明,[C4MIM]Br、[C6MIM]Br和[C8MIM]Br 3種IL對兩種目標物的提取能力順序為C8>C4>C6,提取效率并未隨著離子液體中陽離子的烷基鏈長度增加而增加。Raj[19]和Sankaran[20]等在研究IL的理化特性時也發現了[C6MIM]Br類似的異常結果(親水性順序為C6≥C4>C8)。另外,Zhang等[21]為提高IL對丙烯酰胺的選擇性制備了含有辛基側鏈的IL([OMIM]Br),并發現具有類似結構的[C8MIM]Br對AA有良好的萃取能力,且其萃取原理為[C8MIM]Br能破壞目標物分子間和分子內的氫鍵并在目標物的羥基質子和IL陰離子之間形成新的氫鍵[22]。由于本實驗需要從油樣品基質中提取極性化合物,所以要求萃取劑IL兼溶于水和油,綜上,選擇[C8MIM]Br進行后續研究。
2.2.2 離子液體體積萃取溶劑的體積直接影響DLLME的萃取效率。本實驗考察了[C8MIM]Br體積(60、90、120、150、180、210 μL)對萃取效果的影響。如圖2所示,在60~180 μL范圍內,AA和5-HMF的萃取回收率分別增大至78.9%和68.9%,這是因為較多的IL可以有效吸收和轉移微波能量[17],有利于萃取的傳質過程;但當萃取劑體積大于180 μL后,回收率開始降低,這是因為分析物從溶液到微滴的擴散速率與兩相之間的界面面積有關[23],隨著萃取溶劑總體積增大,溶劑在樣品溶液中的擴散性可能會變差,有些溶劑無法很好地分散到樣品溶液中,導致萃取回收率降低。另一方面,當離子液體體積為180 μL時萃取達到平衡。根據以上結果,選擇180 μL [C8MIM]Br進行后續研究。
2.2.3 微波條件相比于常規DLLME方法,微波輔助可以促使IL和樣品充分混合均勻。由于微波能量會顯著影響目標化合物與IL之間的分子相互作用,因此需要對微波功率、微波輔助萃取時間和溫度進行優化以實現更好的萃取效果。微波功率太低,IL不能很好地分散到樣品溶液中,而微波功率過高則可能會導致分析物的降解。適當地提高萃取溫度會降低IL的粘度,提高IL在油中的溶解性和分散性,從而有助于更好地形成乳濁液。隨著微波功率從200 W增加到300 W,萃取效率迅速增加,當微波功率高于300 W時,萃取效率略有下降。因此,選擇300 W的微波功率。通過考察不同的微波輔助萃取時間發現,在2 ~12 min萃取時間內,目標分析物的回收率逐漸增加,當萃取時間超過12 min后回收率降低。因此,選擇12 min為最佳萃取時間。
適當的高溫有利于IL的分散和萃取傳質,通過考察不同溫度對萃取效果的影響發現,在25~70 ℃溫度范圍內,萃取回收率隨著微波溫度的升高而快速增長,回收率從31.0%增加至85.0%;繼續升溫后萃取回收率開始下降,可能是因為溫度過高會導致樣品基質發生變化,目標物熱分解等。因此,選擇70 ℃為最佳萃取溫度。
2.2.4 樣品油體積在DLLME過程中,樣品體積是影響萃取效果最重要的研究參數之一。在控制加標油濃度不變的條件下,考察了不同大豆油體積(0.5、1、2、3、4 mL)對萃取效果的影響,發現萃取回收率隨著油樣體積從0.5 mL 到2 mL逐漸增加,當樣品體積為2 mL時,AA和5-HMF的萃取回收率分別為95.8%和89.6%;當樣品體積大于2 mL后,隨著樣品體積的增長,萃取回收率開始略微下降,這可能由于,一方面受樣品中復雜基質的影響,另一方面,萃取已逐漸達到平衡,因此選擇2 mL為最佳樣品體積。
2.3 方法評估
通過在空白大豆油中加入不同濃度的混標,制成一系列濃度梯度的加標油,然后在優化條件下以峰面積(y)對加標油的質量濃度(x,μg/mL)制作工作曲線。所建方法的工作曲線、檢出限、定量下限、相關系數和相對標準偏差見表1。兩種目標物在線性范圍0.5~50 μg/mL內呈現良好的線性關系(r2≥0.997 1),AA和5-HMF的檢出限(LOD,S/N=3)分別為0.11、0.70 μg/mL,定量下限(LOQ,S/N=10)分別為0.33、2.33 μg/mL。對樣品進行5、10、 20 μg/mL 3個水平的加標回收實驗,每個水平平行6次,兩種目標物的日內、日間相對標準偏差(RSD)均不大于4.3%,表明方法精密度良好,滿足分析測定要求。

表1 方法的線性回歸方程、檢出限、定量下限、相關系數和相對標準偏差Table 1 Linear equations,LODs,LOQs,correlation coefficients and relative standard deviations(RSDs) of the proposed method
2.4 方法對比
將本方法與近年報道的食品中AA和5-HMF的分析方法進行了對比,見表2。與本方法類似的使用毛細管電泳檢測AA的3種方法[12,24-25]中,均或多或少地使用了有機溶劑(如正己烷、甲醇、乙腈),且前處理包括研磨、液液萃取、脫脂除蛋白和旋蒸濃縮等復雜步驟,不僅前處理時間長,而且液液萃取-毛細管電泳(LLE-CE)[12]和基質固相分散-毛細管電泳(MSPD-CE)[25]兩方法的LOD分別大致高出本方法2倍和5倍;雖然同時測定蜂蜜和植物油中呋喃化合物的膠束電動色譜法[7]的回收率較高,但其在液液萃取過程中也使用了正己烷。相比之下,本方法首次使用無分散劑微波輔助離子液體萃取食用油中AA和5-HMF,且使用了更環保的IL作為萃取劑,未使用有機溶劑,同時各種溶液和試劑的消耗更少,從而更綠色環保,大大降低了實驗成本;本實驗目標物AA的分析速度比馮軍等[12]的快一倍,且更簡單易操作,樣品前處理僅需30 min,兩個目標物在12 min內即可完全分離。

表2 本方法與其他方法的比較Table 2 Comparison of the proposed method with other methods
2.5 實際樣品分析
為評估本方法的實用性,在最佳條件下測試了4種商品食用香油、1種餐館自制花椒油、1種小吃攤煎炸老油、1份廢棄餐廚油粗制品共7個實際樣品中的AA和5-HMF。結果(表3)表明:在3個品牌芝麻香油和餐館自制花椒油中檢出5-HMF,質量濃度分別為3.3、2.2、1.1、1.5 μg/mL,且是首次在花椒油中檢出5-HMF。其他油樣未檢出目標分析物。通過測定3個濃度水平(5、10、20 μg/mL)的加標大豆油,得到AA和5-HMF的加標回收率分別為82.5%~96.2%和77.9%~95.9%,RSD分別為1.3%~5.2%和1.7%~4.9%。圖3為兩種實際樣品及其在10 μg/mL加標水平下的電泳圖。
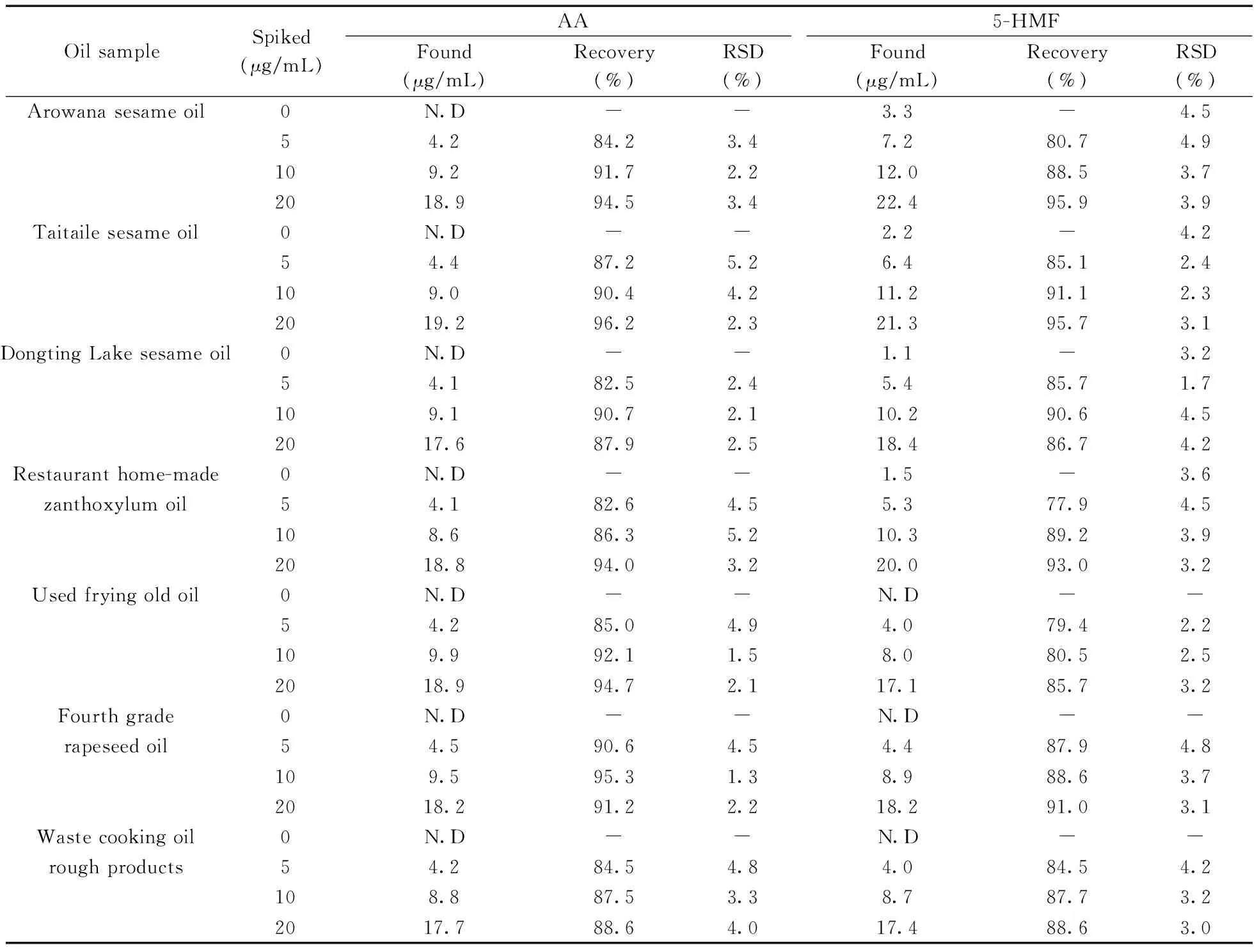
表3 兩種目標物在實際樣品中的含量、加標回收率和相對標準偏差(n=3)Table 3 Concentrations,spiked recoveries and RSDs of the two analytes in the actual samples(n=3)
“N.D” :not detected or below limit of detection
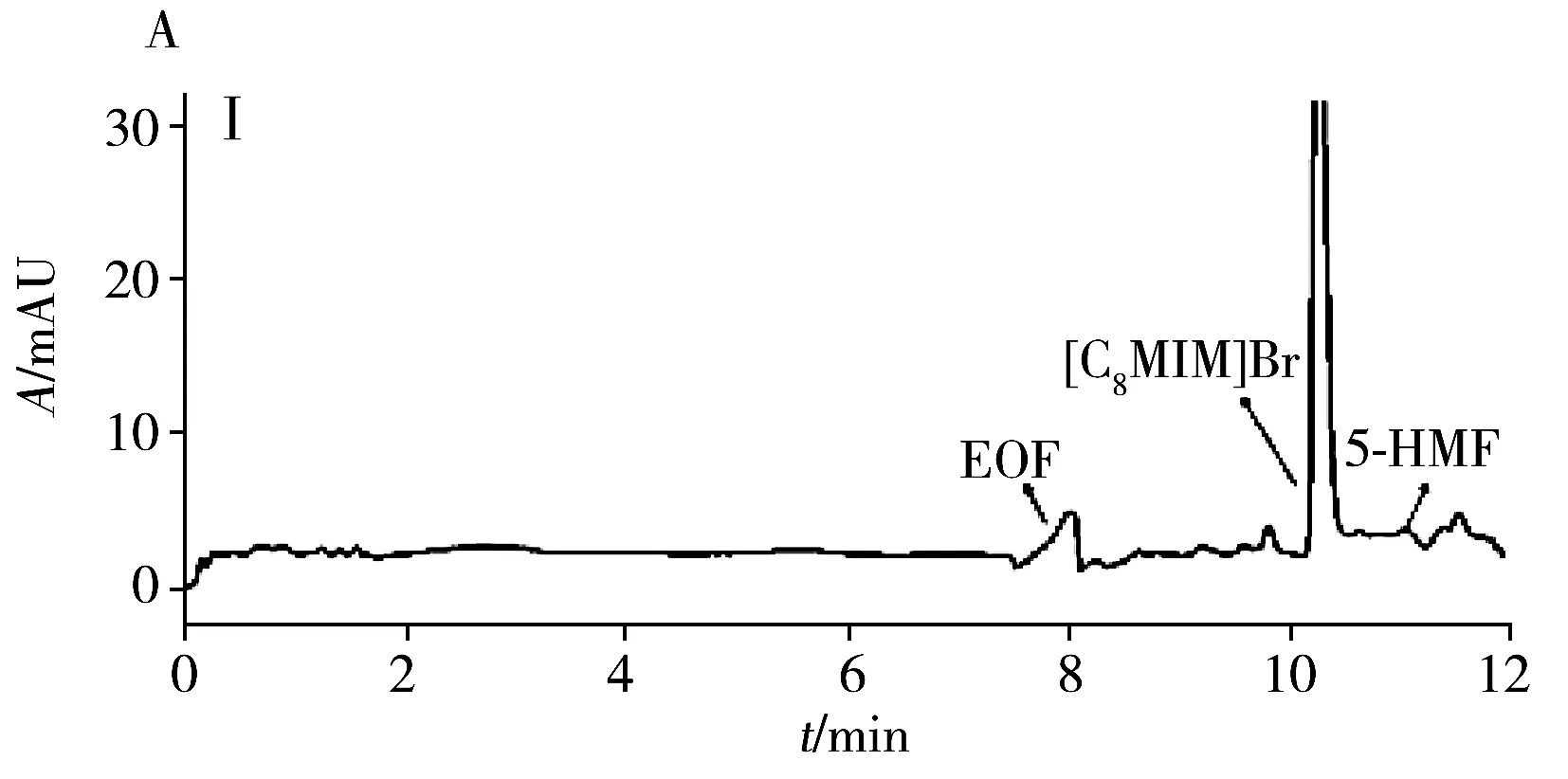
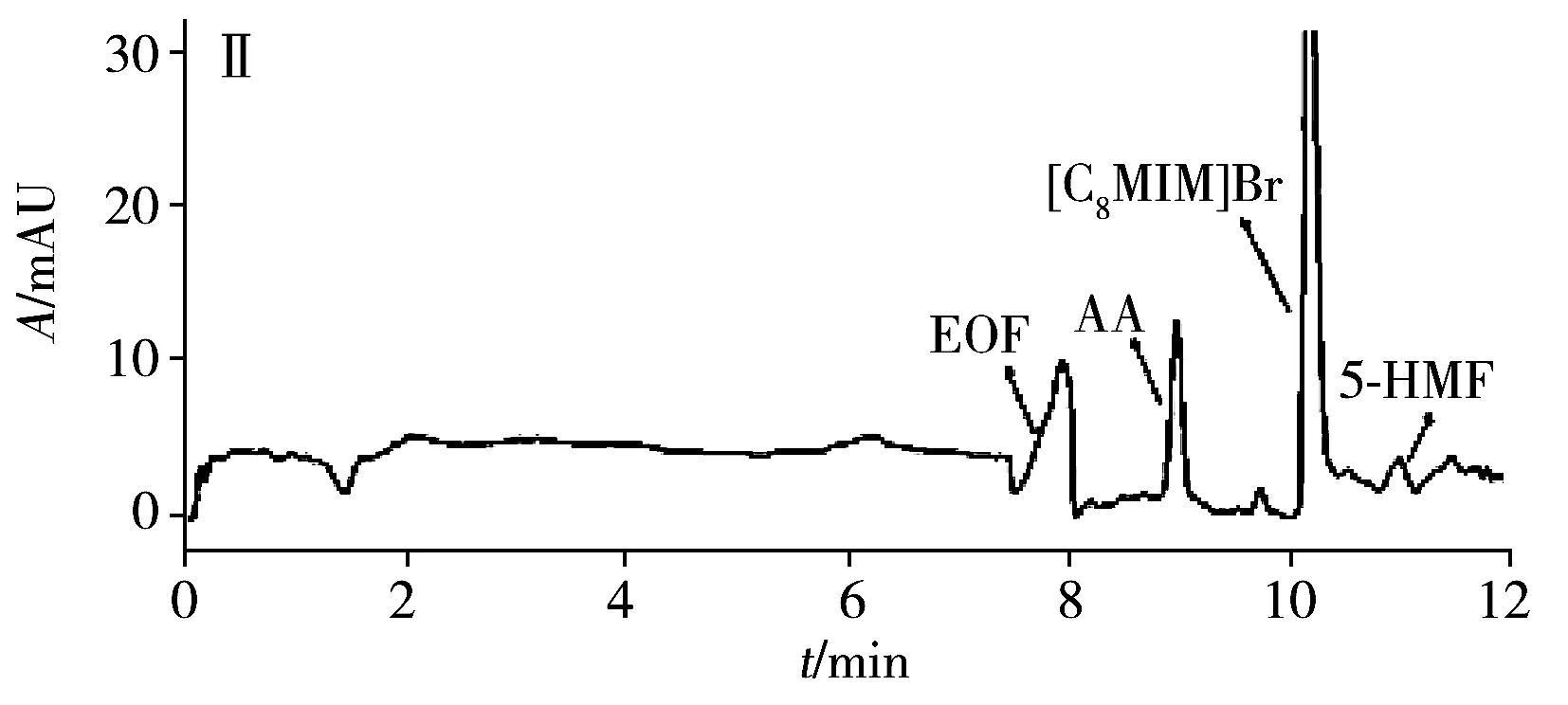
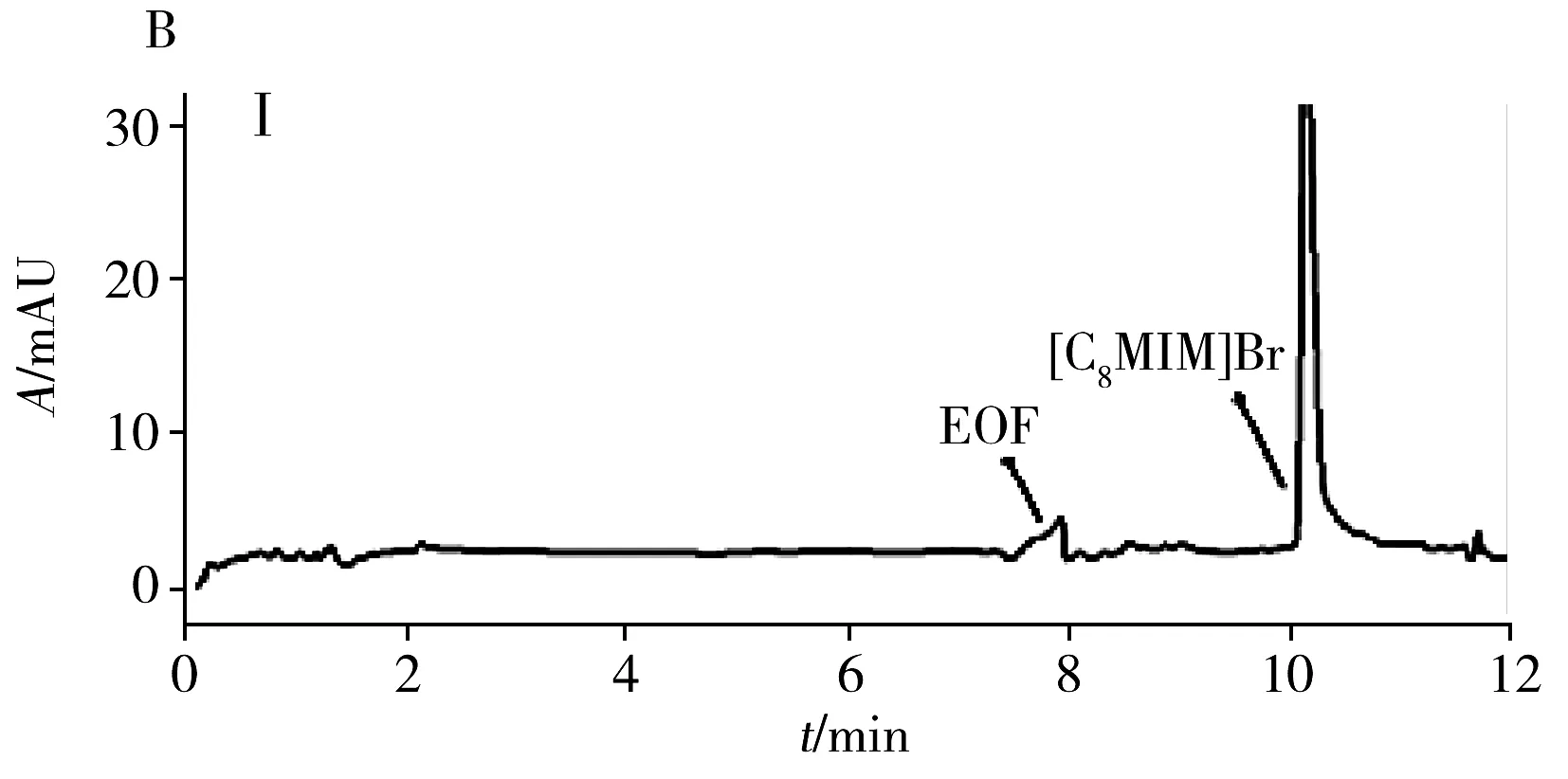
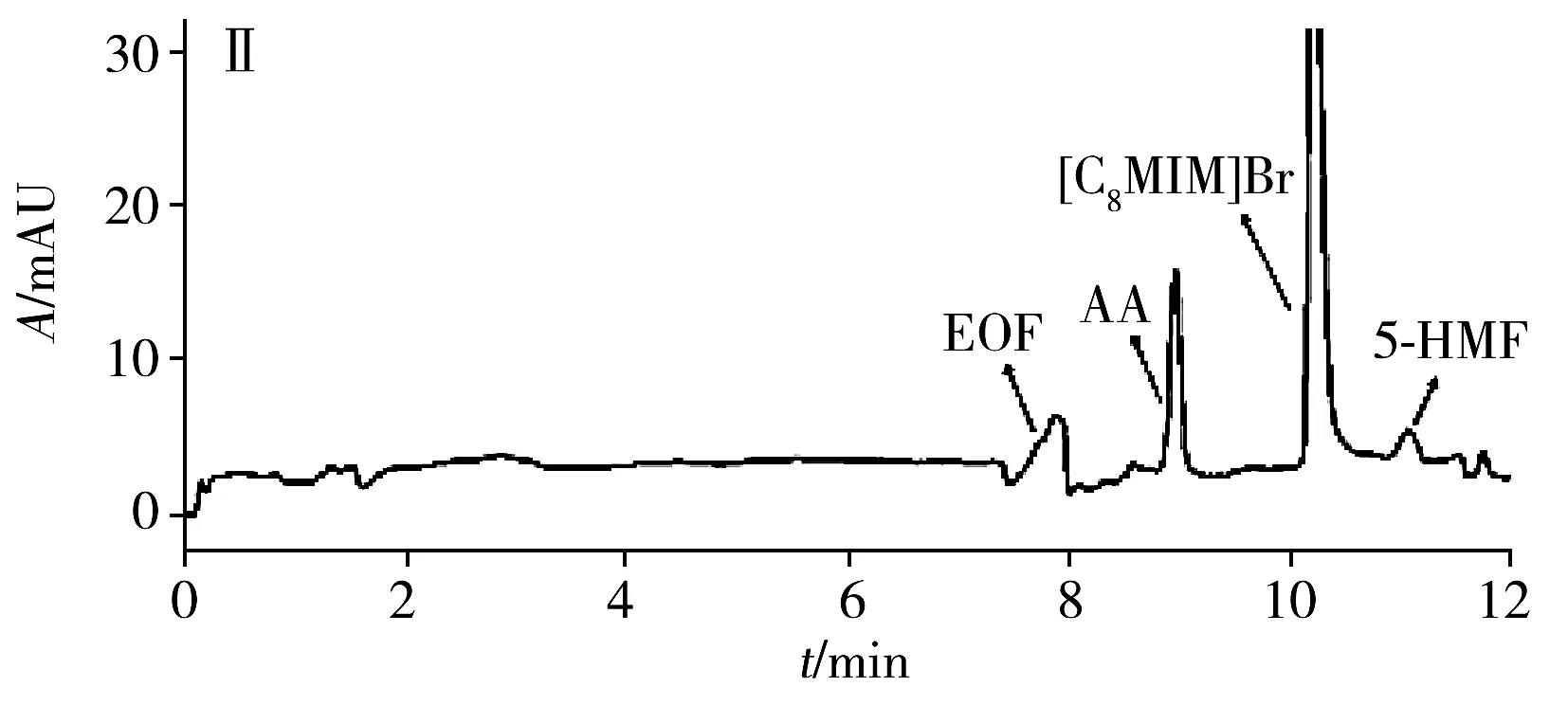
圖3 兩種實際樣品及其加標樣品電泳圖Fig.3 Electropherograms of two actual samples and spiked samplesA:arowana sesame oil;B:used frying old oil;Ⅰ:unspiked sample;Ⅱ:sample spiked with 10 μg/mL
3 結 論
本研究結合IL、無分散溶劑的DLLME、MA以及CE的優勢,開發了一種無需分散劑的微波輔助離子液體分散液液微萃取結合膠束電動色譜分析食用油中AA和5-HMF的方法。方法的加標回收率和相對標準偏差分別為77.9%~96.2% 和1.3%~5.2%,準確性和重現性良好,而且更簡單、快速、高效、低成本和綠色環保。利用此方法在實際樣品芝麻香油和花椒油中檢出5-HMF,含量范圍為1.1~3.3 μg/mL,說明本方法適用于食用油中AA和5-HMF的分析測定。本研究對于食用油質量管控、食用油安全和營養健康等有重要的實際意義。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
