LRG1通過抑制p38/MAPK通路的活化緩解Aβ1-42誘導SH-SY5Y細胞損傷
2020-07-07 07:59:22薛延莉
中風與神經疾病雜志 2020年6期
康 濤, 韓 征, 薛延莉, 楊 謙
阿爾茨海默病(Alzheimer’s disease,AD)是全世界老年人癡呆的主要原因。盡管用于AD預防和治療藥物的研究很多,但該病目前還沒有有效的治療方法,在個人、醫學和社會經濟水平上造成越來越大的負擔[1]。富亮氨酸α2糖蛋白1(Leucine-rich-alpha-2-glycoprotein1,LRG1)是富含亮氨酸重復序列(leucine rich repeat,LRR)蛋白家族中高度保守的成員,參與蛋白間相互作用、信號轉導、細胞粘附、細胞存活、細胞凋亡和細胞轉移等[2,3]。LRG1與各種類型的癌癥、早期中性粒細胞分化、炎性疾病、心衰以及免疫疾病有關[4]。此外,有證據表明LRG1與神經退行性疾病[5]以及特發性正常腦積水[6]有關。Jin等研究發現LRG1加重小鼠腦缺血誘導的損傷[7]。在之前的研究報道LRG1在AD中高表達[8],并且Se-Met作用于小鼠AD模型可以誘導LRG1表達水平下降[9]。然而,LRG1在AD發展中的具體作用仍是未知的。因此,本研究旨在探討LRG1對Aβ1-42誘導SH-SY5Y細胞損傷的作用,以期為AD的治療提供一定的理論依據。
1 材料與方法
1.1 主要試劑 Aβ1-42(美國Sigma公司),人骨髓神經母細胞瘤細胞株(SH-SY5Y,美國ATCC),胎牛血清和DEME(美國Gibco公司),Lipofectamine 2000 轉染試劑盒(美國賽默飛世爾科技公司),Trizol試劑(美國Invitrogen公司),CCK-8(日本Dojindo Molecular Technologies公司),RNA逆轉錄試劑盒、cDNA合成試劑盒和qPCR定量試劑盒(日本Takara公司),qPCR引物(上海生工公司),BCA蛋白濃度測定試劑盒(美國Thermo Scientific公司),SDS-PAGE凝膠配制試劑盒(上海碧云天生物技術公司),聚偏氟乙烯(PVDF)膜(美國Pall Corporation公司),抗LRG1抗體、抗Bcl-2抗體、抗Bax抗體、抗p-p38抗體和抗p38抗體(美國Millipore公司),抗NADPH抗體(美國Sigma公司),辣根過氧化物酶(HRP)二抗(南京鐘鼎生物公司),Annexin V-FITC/PI雙染試劑盒(南京凱基生物科技公司)。
1.2 方法
1.2.1 細胞培養 SH-SY5Y細胞培養于添加了10%熱滅活胎牛血清和0.1%青霉素/鏈霉素的DMEM培養,將細胞置于37 ℃含5% CO2的培養箱中。
1.2.2 實驗分組 (1)使用不同濃度的Aβ1-42(3、6、9、12和15 μmol)處理SH-SY5Y細胞48 h。通過CCK-8實驗檢測細胞活性,Western blot實驗檢測LRG1蛋白表達。(2)SH-SY5Y細胞分為未處理組、Aβ1-42組、Aβ1-42+shRNA組、Aβ1-42+shLRG1組。培養48 h后,通過RT-qPCR檢測LRG1 mRNA表達,CCK-8實驗檢測細胞活性,流式細胞術檢測凋亡率,Western blot實驗檢測凋亡相關蛋白和p-p38的表達情況。(3)SH-SY5Y細胞分為未處理組、Aβ1-42組、Aβ1-42+shLRG1組、Aβ1-42+shLRG1+U-46619組。培養48 h后,通過CCK-8實驗檢測細胞活性,流式細胞術檢測凋亡率。
1.2.3 細胞活性檢測 將SH-SY5Y細胞接種于96孔板,每孔細胞數為1×104,待SH-SY5Y細胞長至85%融合后,分別用(3、6、9、12和15 μmol )Aβ1-42處理SH-SY5Y細胞48 h,不做任何處理的細胞作為空白對照。處理完成后,使用CCK-8試劑盒進行細胞活性檢測。
1.2.4 轉染干擾質粒 將SH-SY5Y細胞接種于96孔板,每孔細胞數為1×104。待細胞進入對數生長期后,按照Lipo2000轉染試劑說明書進行shLRG1和shRNA轉染。以LRG1 mRNA為靶點的慢病毒介導的短發夾RNA(shRNA)合成于上海吉碼公司。序列如下:sense 5’-AGC TAA AAA GAT GTT TTC CCA GAA TGA CTC TCT TGA AGT CAT TCT GGG AAA ACA TCG GG-3’;antisense 5’-AGC TAA AAA TTC TCC GAA CGT GTC ACG TTC TCT TGA AAC GTG ACA CGT TCG GAG AAG GG-3’。
1.2.5 RT-qPCR 未處理組、Aβ1-42、Aβ1-42+shRNA、Aβ1-42+shLRG1細胞均勻接種于96孔板,37 ℃條件下培養48 h。收集各組細胞,Trizol法提取總RNA,取2 μg總RNA進行反轉錄實驗,并根據qPCR試劑盒說明書進行SH-SY5Y細胞LRG1轉錄水平檢測。GAPDH作為內參基因。LRG1定量序列如下:sense 5’-TACAGCACCTGGATATGTTGGA-3’;antisense 5’-GTTGTGGGAGATGTCG-3’。2-ΔΔCT法計算基因的相對表達量,每個樣本設置3個重復。
1.2.6 Western blot 未處理組、Aβ1-42、Aβ1-42+shRNA、Aβ1-42+shLRG1細胞均勻接種于96孔板,37 ℃條件下培養48 h。收集并裂解細胞,BCA法檢測蛋白的總濃度,10%~12% SDS-PAGE凝膠分離蛋白并轉移到PVDF膜上,室溫封閉3 h。用LRG1(1∶100)、Bcl-2(1∶100)、Bax(1∶500)、p-p38(1∶1000)、p38(1∶1000)和NADPH(1∶2000)的初抗4 ℃孵育過夜。隨后,室溫下用HRP二抗(1∶5000)孵育1 h。實驗重復3次。
1.2.7 細胞凋亡檢測 未處理組、Aβ1-42、Aβ1-42+shRNA、Aβ1-42+shLRG1細胞均勻接種于96孔板,37 ℃條件下培養48 h。使用Annexin V-FITC/PI雙染試劑盒檢測各組細胞的凋亡情況。
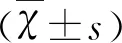
2 結 果
2.1 LRG1在Aβ1-42處理的SH-SY5Y中表達量上調 本研究將SH-SY5Y與Aβ1-42孵育模擬AD的細胞模型。不同濃度的Aβ1-42(3、6、9、12和15 μmol)處理SH-SY5Y細胞48 h后通過CCK-8實驗檢測細胞活性。CCK-8實驗結果表明,與未處理組相比,隨著Aβ1-42的濃度的增加,細胞的活性不斷被抑制(P<0.05,見圖1A)。另外,Western blot實驗結果表明,Aβ1-42處理SH-SY5Y細胞48 h后LRG1蛋白表達水平顯著升高且與Aβ1-42處理濃度有關(P<0.05,見圖1B)。本實驗選取12 μmol作為Aβ1-42體外誘導SH-SY5Y損傷的濃度。
2.2 LRG1沉默緩解Aβ1-42誘導的細胞凋亡 為了探究LRG1對Aβ1-42誘導的細胞損傷的作用,本研究使用LRG1干擾質粒轉染SH-SY5Y細胞。將SH-SY5Y細胞分為以下4組:未處理組、Aβ1-42組、Aβ1-42+shRNA組、Aβ1-42+shLRG1組,培養48 h。通過RT-qPCR檢測實驗LRG1轉錄水平。RT-qPCR結果表明,LRG1沉默顯著抑制Aβ1-42誘導的LRG1轉錄水平(P<0.05,見圖2A)。相似地,使用CKK-8、流式細胞術和Western blot分別檢測細胞活性、凋亡率和凋亡相關蛋白水平。實驗結果發現Aβ1-42處理SH-SY5Y細胞顯著降低細胞活性、提高細胞凋亡率、抑制Bcl-2蛋白水平以及促進Bax蛋白表達;然而LRG1沉默緩解了由Aβ1-42引起的細胞活性的降低、細胞凋亡率的升高、Bcl-2蛋白水平的下調以及Bax蛋白表達的上調(P<0.05,見圖2B~D)。
2.3 LRG1沉默調節p38/MAPK信號通路 為了探究LRG1敲除緩解Aβ1-42誘導SH-SY5Y細胞損傷的可能機制,我們進一步研究了p38/MAPK信號通路。將SH-SY5Y細胞分為以下4組:未處理組、Aβ1-42組、Aβ1-42+shRNA組、Aβ1-42+shLRG1組,培養48 h。通過Western blot檢測實驗p38磷酸化水平。Western blot結果表明,Aβ1-42誘導p38磷酸化水平上調,然而,LRG1沉默抑制Aβ1-42誘導的p38磷酸化水平上調(P<0.05,見圖3A)。因此,LRG1沉默可能抑制Aβ1-42誘導的p38/MAPK信號通路的激活。
2.4 p38/MAPK信號通路的激活減弱LRG1沉默對Aβ1-42誘導細胞損傷的保護作用 隨后,我們檢測了p38/MAPK信號通路和LRG1在Aβ1-42誘導細胞損傷中的關系。將SH-SY5Y細胞分為以下4組:未處理組、Aβ1-42組、Aβ1-42+shLRG1組、Aβ1-42+shLRG1+U-46619組,培養48 h。通過CCK-8和Western blot檢測細胞活性和凋亡率。結果表明,沉默LRG1緩解了由Aβ1-42引起的細胞活性的降低和細胞凋亡率的升高。然而,U-46619(p38信號通路的特異性激活劑)處理顯著抑制了LRG1沉默對Aβ1-42誘導細胞損傷的保護作用(P<0.05,見圖4A、B)。這些結果表明了LRG1沉默保護Aβ1-42誘導的SH-SY5Y細胞損傷是通過抑制p38/MAPK信號通路的激活。

圖1 Aβ1-42對SH-SY5Y細胞活性和LRG1表達的影響與對照組比較*P<0.05




3 討 論
本實驗探究了LRG1在Aβ1-42誘導的SH-SY5Y細胞損傷中的作用和潛在機制。在這里,我們發現Aβ1-42處理可有效地抑制SH-SY5Y細胞活性。此外,Aβ1-42可上調LRG1的表達量,且其表達量的變化與Aβ1-42處理的濃度有關。與本研究結果類似,先前的研究也表明在七氟醚誘導AD小鼠中LRG1水平顯著上調[10],并且Zou等的報道同樣指出LRG1在AD患者中的基因水平顯著高于正常人[8]。研究表明,在心機梗死的小鼠中沉默LRG1促進成纖維細胞的增殖能力[11]。Jin等的研究表明在小鼠急性缺血性腦卒中LRG1過表達顯著促進神經元細胞的凋亡[7]。同樣的,本研究也發現沉默LRG1可有效緩解Aβ1-42誘導的神經細胞活性降低和細胞凋亡增加。細胞凋亡是神經退行性疾病和神經系統疾病的重要原因,如帕金森病和AD[12]。而研究已經證明Aβ積累誘導的神經細胞凋亡在AD發病機制中起著舉足輕重的作用[13]。最近的研究表明在小鼠急性缺血性腦卒中LRG1過表達顯著促進神經元細胞的凋亡和自噬[7]。本研究的結果表明,Aβ1-42處理可以顯著降低SH-SY5Y細胞活性,促進細胞凋亡,而沉默LRG1逆轉了Aβ1-42的作用。這些結果表明LRG1沉默可以緩解Aβ1-42誘導的SH-SY5Y細胞損傷。
絲裂原活化蛋白激酶(mitogen-activated protein kinases,MAPKs)在細胞外刺激轉化為一系列胞內磷酸化級聯過程中起著重要的轉導作用[14]。研究表明MAPKs調節Aβ1-42誘導AD神經炎癥反應、神經元死亡和認知缺失[15]。p38信號途徑作為MAPKs中一種,優先被炎癥細胞因子和細胞外壓力(如紫外線、熱量和活性氧)激活[16]。在早期AD患者的腦部可以發現p38/MAPK信號的激活,而減弱p38/MAPK信號可以減少Aβ1-42誘導神經元死亡[17]。Ban等研究表明在甲狀腺癌細胞中過表達LRG1顯著增加p38的磷酸化水平[18]。另外,Xie等的結果也證明了LRG1與表皮生長因子受體相互作用進一步激活了p38/MAPK信號通路[19]。本研究結果表明,沉默LRG1可以抑制Aβ1-42誘導的p38的磷酸化和細胞凋亡,并且p38/MAPK信號通路的特異性激活劑U-46619可以逆轉保護效果。這些結果表明LRG1沉默可以抑制p38/MAPK信號通路的激活從而緩解Aβ1-42誘導的細胞損傷。
綜上所述,LRG1沉默可以緩解Aβ1-42誘導的SH-SY5Y細胞損傷。其機制可能是LRG1沉默抑制p38/MAPK信號通路的激活。因此,LRG1可以作為一個潛在的分子靶點用于治療老年癡呆。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
電子制作(2018年11期)2018-08-04 03:25:42
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
