柱前衍生—液質聯用法快速測定葡萄中單氰胺殘留
2020-07-30 01:32:50占繡萍黃蘭淇陳建波朱衛芳
農藥科學與管理 2020年3期
關鍵詞:檢測
占繡萍,陳 秀,黃蘭淇,馬 琳,陳建波,朱衛芳,趙 莉
(1.上海市農業技術推廣服務中心,農業農村部農藥質量監督檢驗測試中心(上海),上海 201103;2.上海市青浦區農業技術推廣服務中心,上海 201799)
單氰胺(cyanamide)是一種特殊的化合物,分子式為CH2N2,它是一種氨基氰。這種氰類化合物普遍都有著比較強的毒性,尤其是單氰胺這一種化合物,有著極強的生物腐蝕性。作為一種毒性較強的化學藥物,單氰胺能夠腐蝕人體皮膚組織,并且刺激呼吸道,以及呼吸道黏膜。一旦單氰胺中毒,就會使中毒者呼吸急促,頭暈頭痛等情況[1-4]。美國環境保護組織EPA將單氰胺列為高毒Ⅰ類危險農藥[5]。
目前在國內市場登記的主要為50%單氰胺水劑,允許登記使用的作物為葡萄,并且不同廠家的產品毒性略不同,分別為低毒或中毒,它的主要作用是刺激葡萄的活性物質,致使新陳代謝所需的核苷酸合成減少,起到打破休眠、促進萌芽、提前成熟等生長調節作用,可促使作物萌動初期芽齊、芽壯,還可增加作物單產,改善品質等。根據本市地產草莓、葡萄使用植物生長調節劑對品質和質量安全的風險評估項目的開展,調查發現單氰胺也是上海葡萄種植戶使用非常普遍的植調劑之一。并且在GB2763-2019 食品安全國家標準 食品中農藥最大殘留限量中也明確規定了葡萄中的最大殘留限量為0.05mg/kg (臨時限量),因此迫切需要建立葡萄中快速、準確的檢測單氰胺的殘留方法。
單氰胺極易溶于水,使用常規有機溶劑難以提取,相對分子量小,濃縮過程易揮發,無特征吸收峰,遇酸堿不穩定,其殘留檢測極具挑戰性。目前,單氰胺的檢測還沒有國家標準和行業標準,關于單氰胺的殘留檢測方法報道也較少,主要以柱前衍生間接測定為主。本文參照一些文獻[6-10]的方法,進行不斷的優化改進,只需進行簡單的衍生處理,無需凈化,即可滿足殘留檢測要求,建立了一種在葡萄中相對簡便、快速、高效、準確的單氰胺殘留檢測方法。
1 實驗部分
1.1 主要儀器與裝置 Agilent1290 UPLC-Agilent6460 MS/MS超高效液相色譜-三重四極桿串聯質譜聯用儀:配ESI源,超聲波(Jump Ultrasonic cleaning),高速勻漿機(IKA T25 digital);GB11240-89電熱恒溫水浴鍋;AC-40氮吹濃縮器;國華SHAC恒溫振蕩器;TDL-5-A離心機;Milli Q超純水系統;漩渦混合器(SCI LoGex MX-S)。
1.2 試劑及溶液的配制 甲醇(色譜純,德國Merck),丙酮(色譜純,TEDIA),甲酸(色譜純,美國Fluka),高純水,氯化鈉(分析純);乙酸乙酯(分析純);單氰胺(純度≥98%)和丹磺酰氯(DNS)標準品(純度≥98%)。
5g/L DNS衍生劑:稱取0.5g DNS,用丙酮溶解至50 mL。
碳酸鈉溶液(0.2mol/L):稱取0.53g碳酸鈉,用水溶解并定容至25mL。
碳酸氫鈉溶液(0.2mol/L):稱取1.68g碳酸氫鈉, 用水溶解并定容至100mL。
碳酸鈉/碳酸氫鈉混合溶液(0.2mol/L,4+46):吸取4 mL 0.2mol/L碳酸鈉水溶液,加入46 mL 0.2 mol/L 碳酸氫鈉水溶液混合均勻。
單氰胺標準工作溶液:準確稱取一定量的單氰胺標準品,用甲醇溶解定容至25 mL,轉移到棕色標準品儲備瓶中保存,然后再稀釋至120 mg/L備用。臨用前根據需要用丙酮稀釋成0.000 3、0.003、0.03、0.15、0.3mg/kg 的單氰胺標準工作溶液。
1.3 樣品前處理方法
1.3.1 提取 準確稱取粉碎后的葡萄樣品5.0g ( 精確至0.01g) ,置于50 mL 塑料離心管中,補水1.0mL,然后加入25 mL 丙酮,并蓋上蓋子,渦旋振蕩3 min,超聲20min,以 4 000r/min 離心3min,待衍生化。
1.3.2 衍生化 葡萄樣品衍生化:取0.5mL 上清液于10mL離心管中,加入0.5mL碳酸鈉/碳酸氫鈉混合溶液,混勻后再加入0.5mL丹磺酰氯丙酮溶液,渦旋1min,于50℃水浴中靜置衍生1h,衍生后,過0.22μm有機濾膜,待測。
單氰胺標準溶液也按照此方法同時進行衍生化。
1.4 檢測條件
1.4.1 色譜條件 色譜柱: Agielnt Eclipse Plus C18柱( 3.0 mm×150mm,3.5μm) ; 以0.1%甲酸水溶液( A 相) 和甲醇(B 相)=50+50為流動相進行等度度洗脫; 流速為0.4mL/min; 進樣量: 2μL; 柱溫: 40℃。目標化合物出峰時間為3.5min左右。
1.4.2 質譜條件 采用AJS ESI源,ESI-電離模式,多反應監測(MRM)檢測;霧化氣壓力:310.3kPa(40psi);干燥氣溫度與流速:300℃,7L/min;鞘氣溫度與流速:350℃,12L/min;毛細管電壓:負離子 3 500V。

表1 單氰胺的質譜分析參數
2 結果與討論
2.1 前處理條件的確定
2.1.1 提取劑的選擇 文獻研究表明,單氰胺在水中溶解度高,但是提取不易濃縮,而且易提取出一些極性物質,影響衍生;單氰胺用乙腈提取,有機層衍生,回收率低,所以本研究不考慮水和乙腈做提取劑。本試驗選取沸點低、對單氰胺有較高溶解度的丙酮、甲醇、乙酸乙酯等3種常用試劑來考察,再通過高速勻漿提取1min,用乙酸乙酯提取的需要加無水硫酸鈉除水后衍生,其他試劑不需除水,直接衍生。結果表明,在丙酮、甲醇、乙酸乙酯3組試驗中,單氰胺回收率均不足50%,回收率具體數據(表2)。原因可能是提取方式、提取時間不夠,導致不能完全提取出該化合物。

表2 葡萄基質中采用不同提取劑時單氰胺的回收率
2.1.2 提取方式的選擇 根據2.1.1中提取劑篩選試驗的結果,本試驗首選用丙酮進行提取方式的進一步優化,勻漿1min、勻漿2min、超聲10min 和超聲20min。結果發現,超聲20min,回收率能夠滿足要求。原因可能是超聲波穿透作用強,加速了單氰胺從基質中萃取到大量溶劑中,而超聲10min,穿透的時間還不夠,導致回收率不夠,所以本試驗最終選擇了自動超聲20min的提取方式。需要注意的是建議使用扣蓋的離心管,并且在管口和蓋子接觸的地方再用保鮮膜封一層,避免因為超聲時間久、溶劑揮發,導致檢測結果不準確。具體結果(表3)。

表3 葡萄基質中不同方式、不同時間提取單氰胺的回收率
2.2 質譜條件優化 本試驗基于三重四級桿串聯質譜儀確立單氰胺衍生物的質譜條件,衍生產物分子量為275,質譜通過MS SCAN方式分別進行正、負離子模式全掃描,調節碎裂電壓,發現正離子模式掃描方式下,可以得到較強的[M+H]峰母離子276,負離子模式掃描方式下,得到較強的[M-H]峰母離子274 。根據子離子掃描、MRM方式,正離子模式下得到豐度較好的2個碎片離子,分別為156.1(定量離子)、171.1(定性離子);負離子模式下得到豐度較好的2個碎片離子,分別為258(定量離子)、230(定性離子)。經過比較,發現負離子模式出峰時間更快,并且靈敏度、峰形略優于正離子模式方式,因此本試驗選擇負離子模式MRM電離方式。0.1mg/kg的色譜圖和碎片圖譜正負離子模式(圖1、2)。
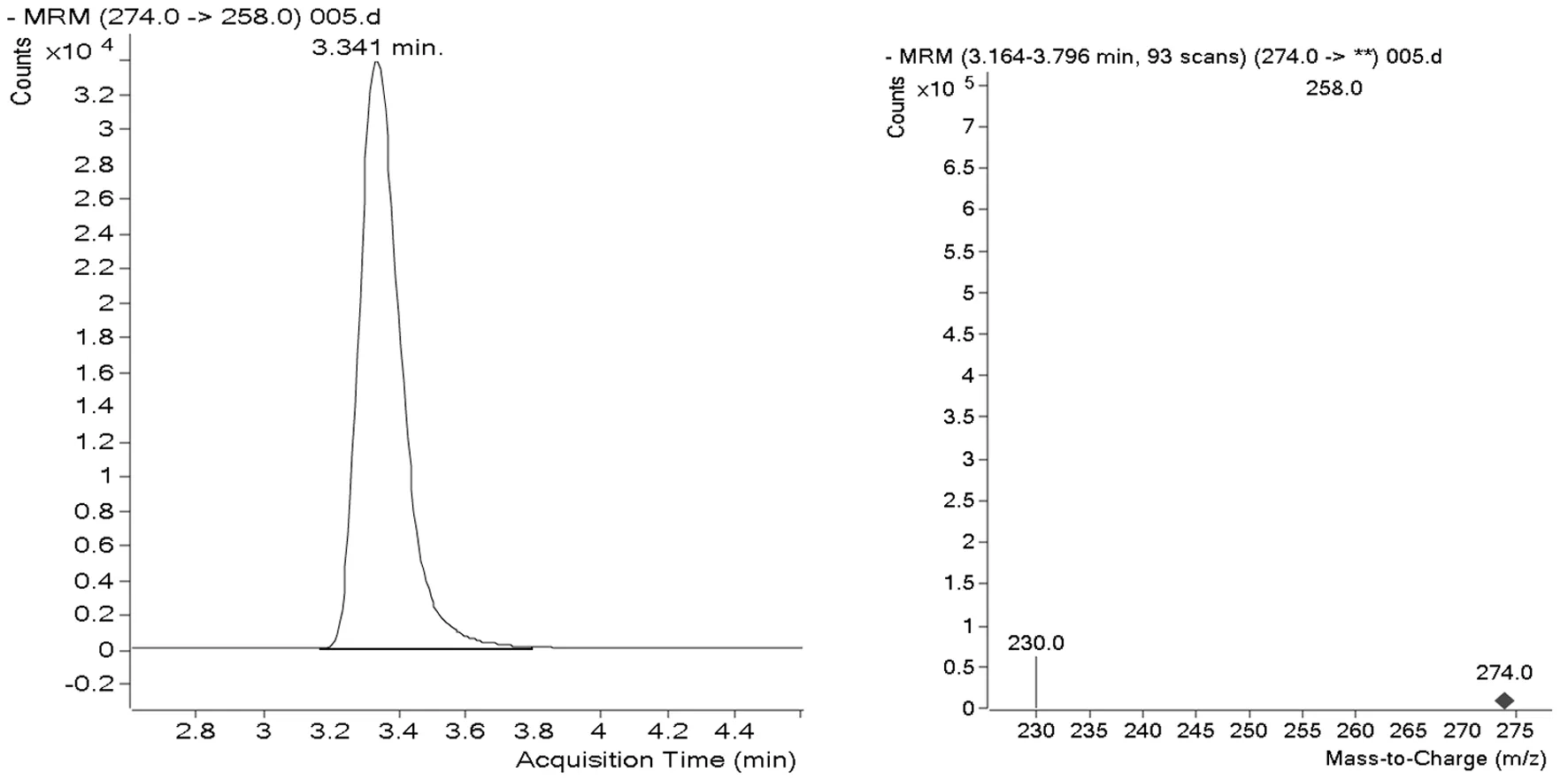
圖1 0.1mg/kg單氰胺的色譜圖、碎片圖(MRM/ ESI-)
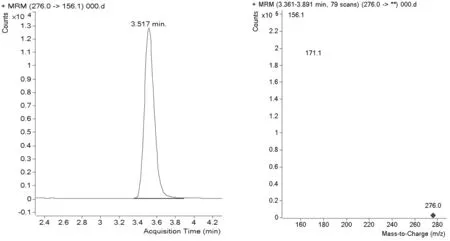
圖2 0.1mg/kg單氰胺的色譜圖、碎片圖(MRM/ ESI+)
2.3 結果計算 試樣中單氰胺的殘留量以質量分數ω計,以mg/kg表示,按公式(1)計算:
(1)
式中:
A——試樣溶液中單氰胺衍生物的峰面積;
As——標準溶液中單氰胺衍生物的峰面積;
ρs——標準工作溶液中單氰胺的質量濃度, mg/L;
V——提前液的總體積(提取有機溶劑體積加補水的體積再加上基質含水的體積),mL;
m——試樣的質量,g;
3——衍生稀釋倍數。
計算結果以重復性條件下獲得的2次獨立測定結果的算術平均值表示,保留2位有效數字,當結果>1mg/kg時,保留3位有效數字。
2.4 回收率、精密度和定量限 按0.001、0.01、0.05、0.1mg/kg 4個添加水平向葡萄樣品中添加單氰胺,每個添加水平重復5次,并做空白對照。按本方法對樣品進行處理和測定,考察單氰胺的回收率,具體結果(表4)。單氰胺在葡萄中的平均回收率為79.7%~92.9 %,相對標準偏差為3.6%~5.1%。可見該方法具有較高的回收率和較好的精密度,可以滿足葡萄中單氰胺殘留量檢測要求。根據添加回收試驗確定葡萄中單氰胺殘留檢測定量限為0.001mg/kg 。
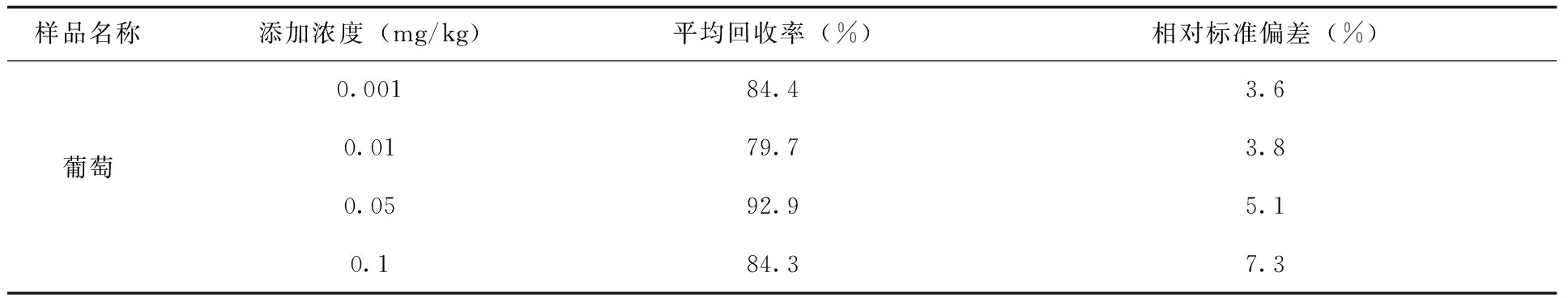
表4 葡萄中單氰胺平均回收率和相對標準偏差(n=5)
2.5 線性方程 取一定量的單氰胺儲備液,用丙酮稀釋成0.000 3、0.003、0.03、0.15、0.3 mg/kg 的標準工作溶液。按照1.3.2衍生化條件進行衍生,實際上機濃度為0.000 1、0.001、0.01、0.05、0.1mg/kg。以標樣的濃度為橫坐標,定量離子的峰面積為縱坐標,繪制成單氰胺衍生物的標準曲線圖,得到線性回歸方程y = 3E+06x + 16 448,R=0.991,線性較好。
2.6 實際樣品測定 按所建立的方法對2019年抽取了上海散戶種植的葡萄中單氰胺殘留量進行測定。20個葡萄樣品中有6個樣品檢測單氰胺殘留,殘留量為0.001 1~0.008 4mg/kg。對照最新發布的GB 2763-2019食品安全國家標準,葡萄中單氰胺MRL為0.05mg/kg(臨時限量),檢出的6個樣品均未超標。
3 結論
本研究利用丙酮超聲提取,無需凈化直接與DNS衍生反應,建立了液相色譜-串聯質譜快速測定葡萄中單氰胺殘留的方法。前處理過程簡便、快速、高效、準確,靈敏度高、線性范圍、準確度和精密度均滿足農藥殘留檢測要求,適用于葡萄樣品中大批量檢測。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
